

Deep learning-driven fragment ion series classification enables highly precise and sensitive de novo peptide sequencing
- PMID: 38167372
- PMCID: PMC10762064
- DOI: 10.1038/s41467-023-44323-7
Deep learning-driven fragment ion series classification enables highly precise and sensitive de novo peptide sequencing
Abstract
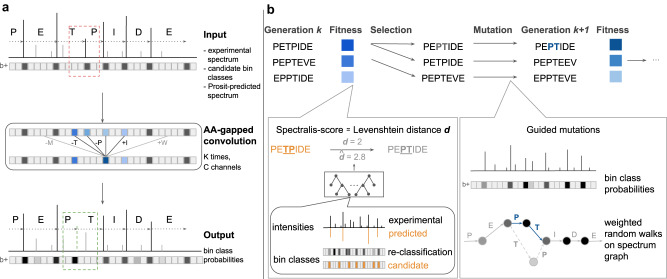
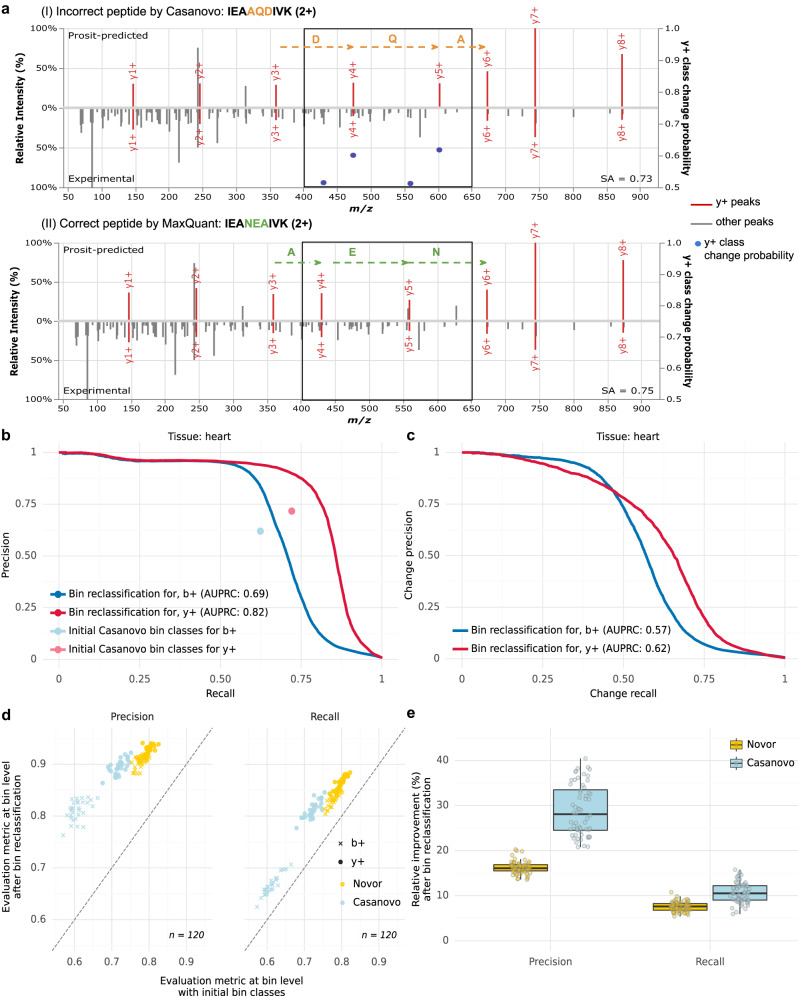
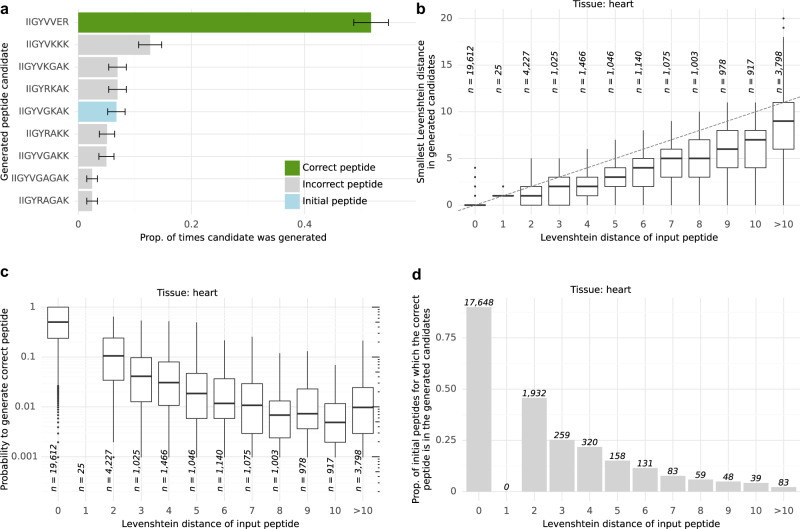
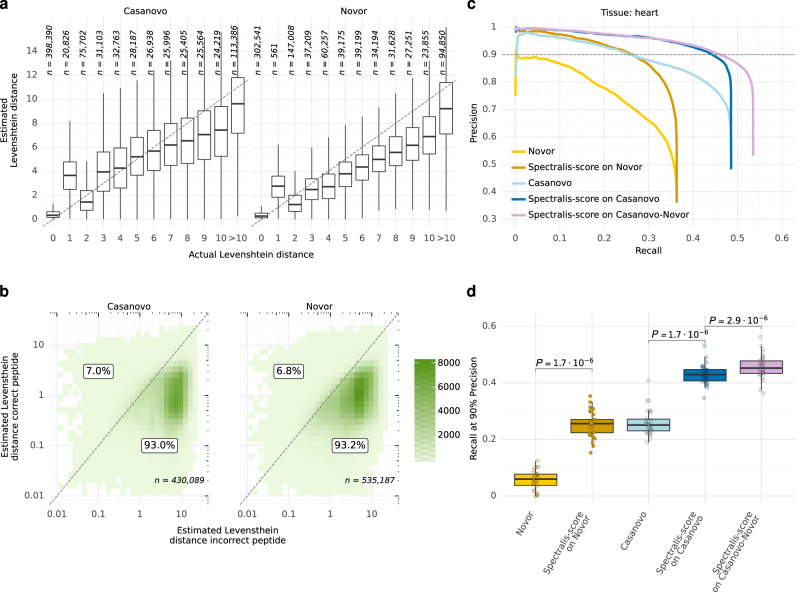
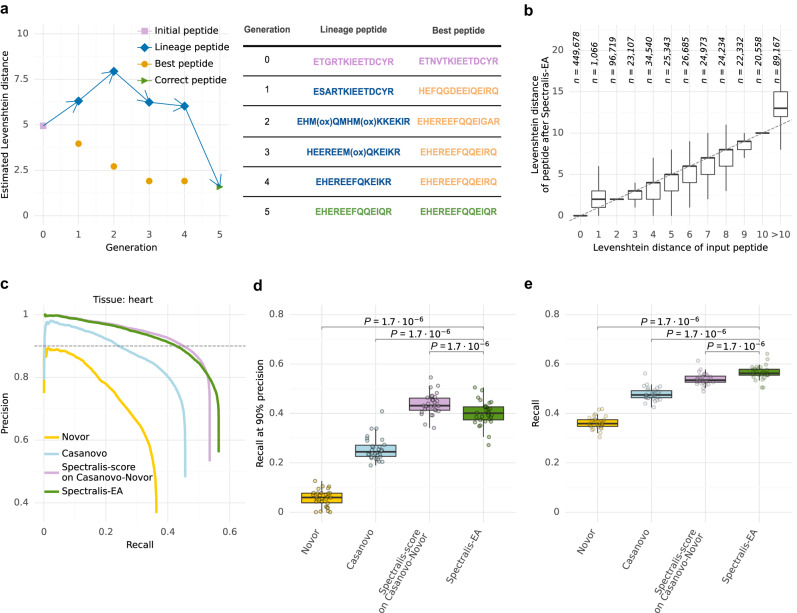
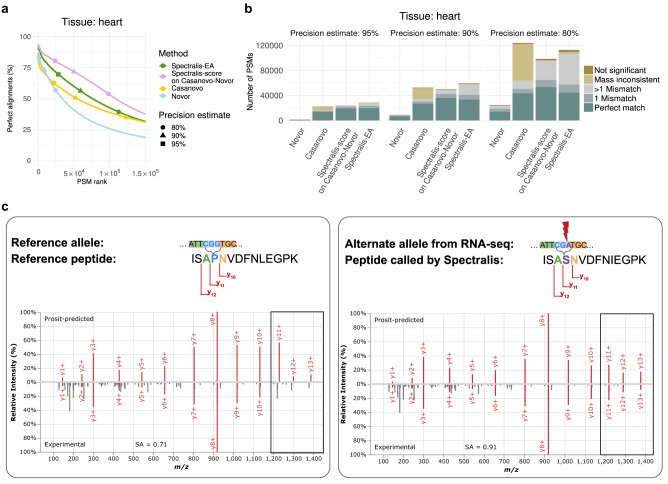
Unlike for DNA and RNA, accurate and high-throughput sequencing methods for proteins are lacking, hindering the utility of proteomics in applications where the sequences are unknown including variant calling, neoepitope identification, and metaproteomics. We introduce Spectralis, a de novo peptide sequencing method for tandem mass spectrometry. Spectralis leverages several innovations including a convolutional neural network layer connecting peaks in spectra spaced by amino acid masses, proposing fragment ion series classification as a pivotal task for de novo peptide sequencing, and a peptide-spectrum confidence score. On spectra for which database search provided a ground truth, Spectralis surpassed 40% sensitivity at 90% precision, nearly doubling state-of-the-art sensitivity. Application to unidentified spectra confirmed its superiority and showcased its applicability to variant calling. Altogether, these algorithmic innovations and the substantial sensitivity increase in the high-precision range constitute an important step toward broadly applicable peptide sequencing.
© 2024. The Author(s).
Conflict of interest statement
M.W. is founder and shareholder of OmicScouts GmbH and MSAID GmbH, with no operational role in either company. The remaining authors declare no competing interests.
Figures
Similar articles
-
Evaluating de novo sequencing in proteomics: already an accurate alternative to database-driven peptide identification?Brief Bioinform. 2018 Sep 28;19(5):954-970. doi: 10.1093/bib/bbx033. Brief Bioinform. 2018. PMID: 28369237
-
Algorithms for de-novo sequencing of peptides by tandem mass spectrometry: A review.Anal Chim Acta. 2023 Aug 8;1268:341330. doi: 10.1016/j.aca.2023.341330. Epub 2023 May 8. Anal Chim Acta. 2023. PMID: 37268337 Review.
-
Application of de Novo Sequencing to Large-Scale Complex Proteomics Data Sets.J Proteome Res. 2016 Mar 4;15(3):732-42. doi: 10.1021/acs.jproteome.5b00861. Epub 2016 Jan 25. J Proteome Res. 2016. PMID: 26743026
-
Application of a Novel Hybrid CNN-GNN for Peptide Ion Encoding.J Proteome Res. 2023 Feb 3;22(2):323-333. doi: 10.1021/acs.jproteome.2c00234. Epub 2022 Dec 19. J Proteome Res. 2023. PMID: 36534699 Free PMC article.
-
Algorithms for the de novo sequencing of peptides from tandem mass spectra.Expert Rev Proteomics. 2011 Oct;8(5):645-57. doi: 10.1586/epr.11.54. Expert Rev Proteomics. 2011. PMID: 21999834 Review.
Cited by
-
A transformer model for de novo sequencing of data-independent acquisition mass spectrometry data.Nat Methods. 2025 Jul;22(7):1447-1453. doi: 10.1038/s41592-025-02718-y. Epub 2025 Jul 1. Nat Methods. 2025. PMID: 40596427
-
De novo peptide databases enable protein-based stable isotope probing of microbial communities with up to species-level resolution.Environ Microbiome. 2025 Aug 26;20(1):111. doi: 10.1186/s40793-025-00767-6. Environ Microbiome. 2025. PMID: 40859350 Free PMC article.
-
π-PrimeNovo: an accurate and efficient non-autoregressive deep learning model for de novo peptide sequencing.Nat Commun. 2025 Jan 2;16(1):267. doi: 10.1038/s41467-024-55021-3. Nat Commun. 2025. PMID: 39747823 Free PMC article.
-
Integration of proteomics profiling data to facilitate discovery of cancer neoantigens: a survey.Brief Bioinform. 2025 Mar 4;26(2):bbaf087. doi: 10.1093/bib/bbaf087. Brief Bioinform. 2025. PMID: 40052441 Free PMC article. Review.
-
A learned score function improves the power of mass spectrometry database search.Bioinformatics. 2024 Jun 28;40(Suppl 1):i410-i417. doi: 10.1093/bioinformatics/btae218. Bioinformatics. 2024. PMID: 38940129 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
