

Distinct patterns of proteostasis network gene expression are associated with different prognoses in melanoma patients
- PMID: 38167612
- PMCID: PMC10761826
- DOI: 10.1038/s41598-023-50640-0
Distinct patterns of proteostasis network gene expression are associated with different prognoses in melanoma patients
Abstract
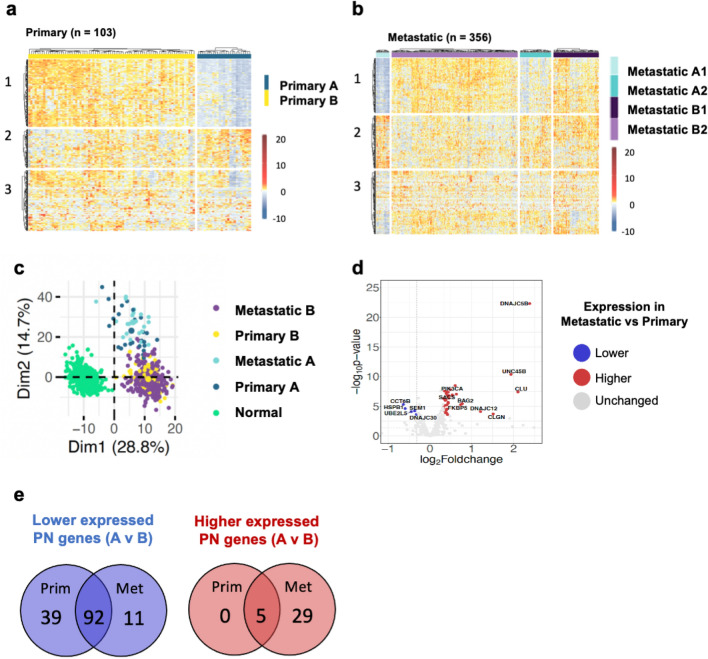
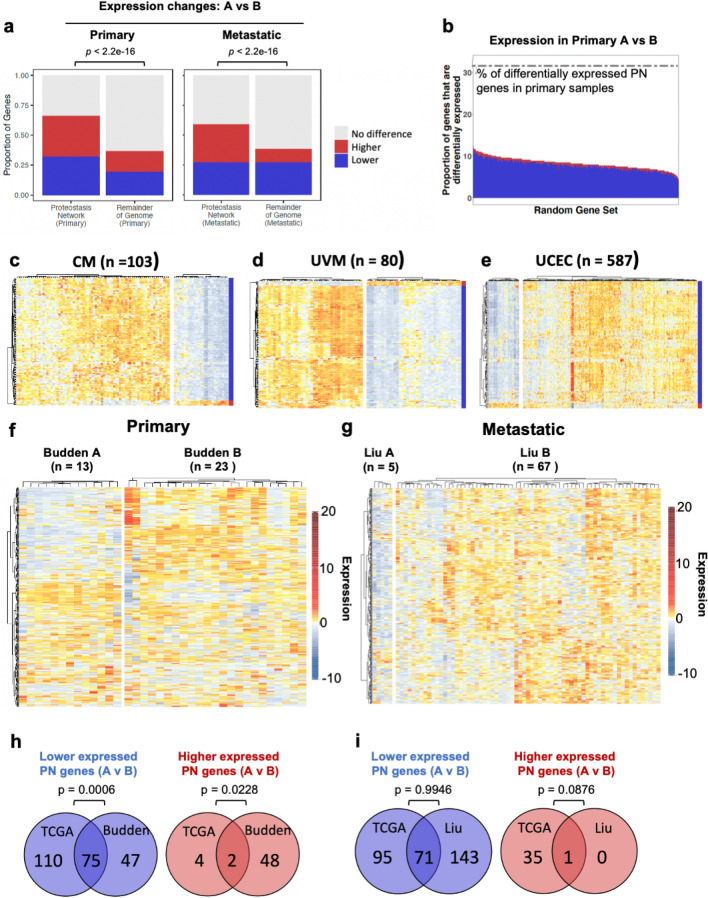
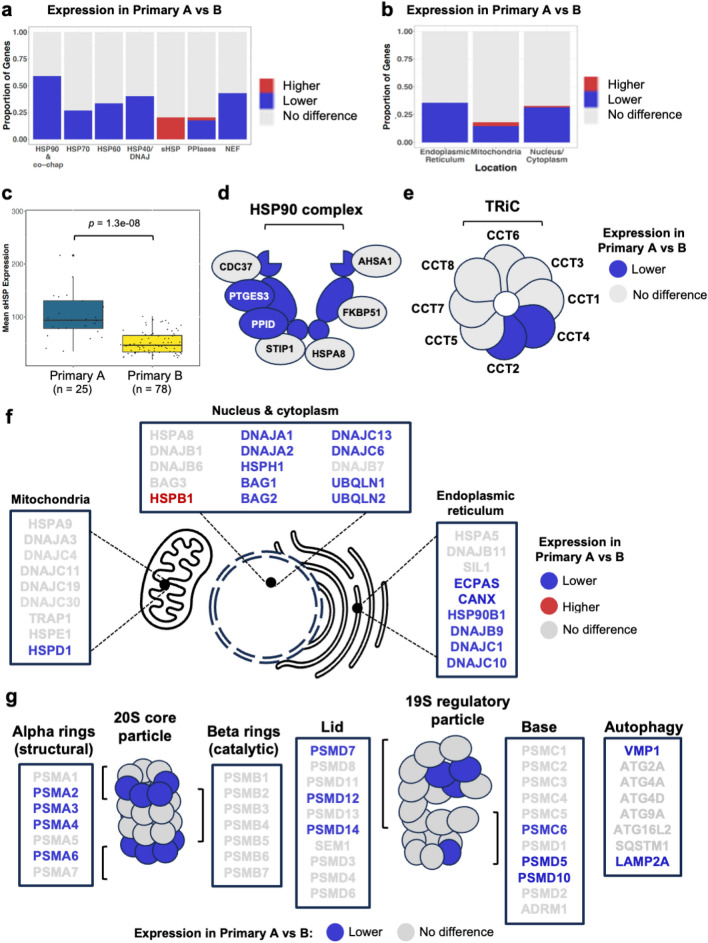
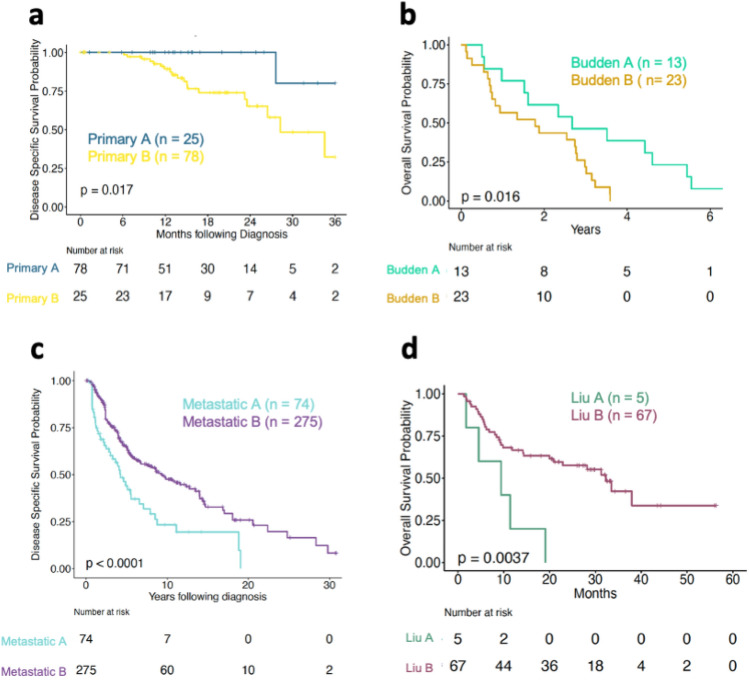
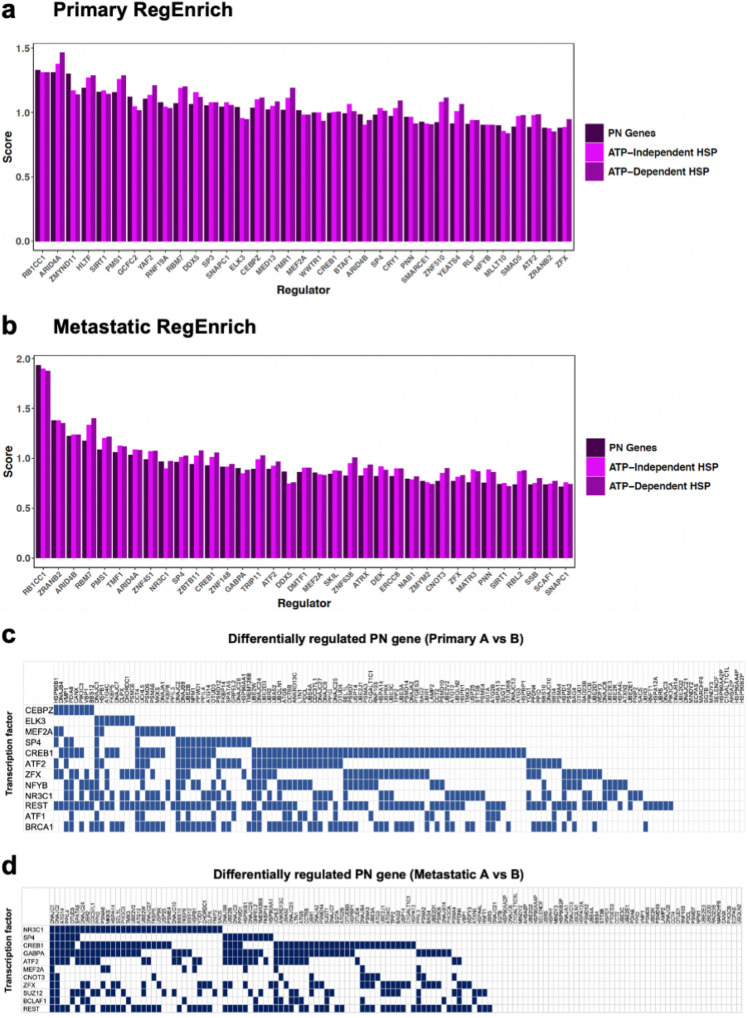
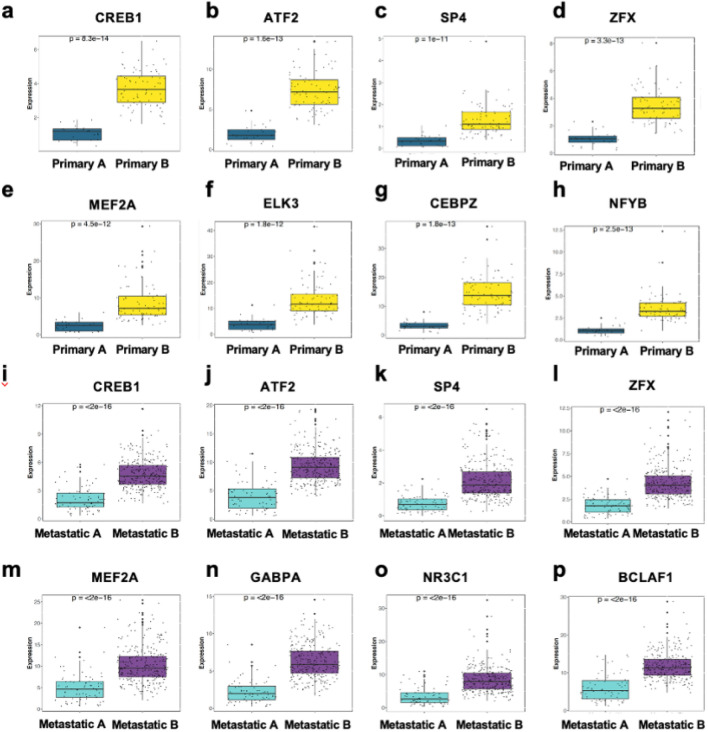
The proteostasis network (PN) is a collection of protein folding and degradation pathways that spans cellular compartments and acts to preserve the integrity of the proteome. The differential expression of PN genes is a hallmark of many cancers, and the inhibition of protein quality control factors is an effective way to slow cancer cell growth. However, little is known about how the expression of PN genes differs between patients and how this impacts survival outcomes. To address this, we applied unbiased hierarchical clustering to gene expression data obtained from primary and metastatic cutaneous melanoma (CM) samples and found that two distinct groups of individuals emerge across each sample type. These patient groups are distinguished by the differential expression of genes encoding ATP-dependent and ATP-independent chaperones, and proteasomal subunits. Differences in PN gene expression were associated with increased levels of the transcription factors, MEF2A, SP4, ZFX, CREB1 and ATF2, as well as markedly different survival outcomes. However, surprisingly, similar PN alterations in primary and metastatic samples were associated with discordant survival outcomes in patients. Our findings reveal that the expression of PN genes demarcates CM patients and highlights several new proteostasis sub-networks that could be targeted for more effective suppression of CM within specific individuals.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
A systematic atlas of chaperome deregulation topologies across the human cancer landscape.PLoS Comput Biol. 2018 Jan 2;14(1):e1005890. doi: 10.1371/journal.pcbi.1005890. eCollection 2018 Jan. PLoS Comput Biol. 2018. PMID: 29293508 Free PMC article.
-
Chemical Biology Framework to Illuminate Proteostasis.Annu Rev Biochem. 2020 Jun 20;89:529-555. doi: 10.1146/annurev-biochem-013118-111552. Epub 2020 Feb 25. Annu Rev Biochem. 2020. PMID: 32097570 Free PMC article. Review.
-
Proteostasis impairment in protein-misfolding and -aggregation diseases.Trends Cell Biol. 2014 Sep;24(9):506-14. doi: 10.1016/j.tcb.2014.05.003. Epub 2014 Jun 16. Trends Cell Biol. 2014. PMID: 24946960 Review.
-
Underlying mechanisms and chemical/biochemical therapeutic approaches to ameliorate protein misfolding neurodegenerative diseases.Biofactors. 2017 Nov;43(6):737-759. doi: 10.1002/biof.1264. Epub 2016 Feb 22. Biofactors. 2017. PMID: 26899445 Review.
-
Alpha 1-Antitrypsin Deficiency: A Disorder of Proteostasis-Mediated Protein Folding and Trafficking Pathways.Int J Mol Sci. 2020 Feb 21;21(4):1493. doi: 10.3390/ijms21041493. Int J Mol Sci. 2020. PMID: 32098273 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
