

Integrated multi-omics analyses identify anti-viral host factors and pathways controlling SARS-CoV-2 infection
- PMID: 38168026
- PMCID: PMC10761986
- DOI: 10.1038/s41467-023-44175-1
Integrated multi-omics analyses identify anti-viral host factors and pathways controlling SARS-CoV-2 infection
Abstract
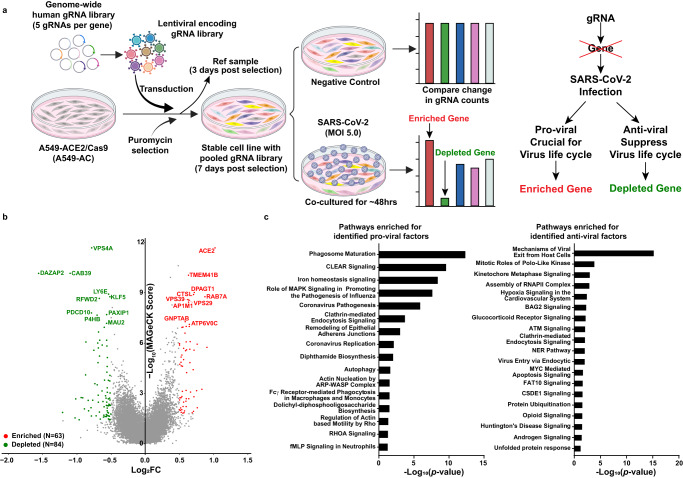
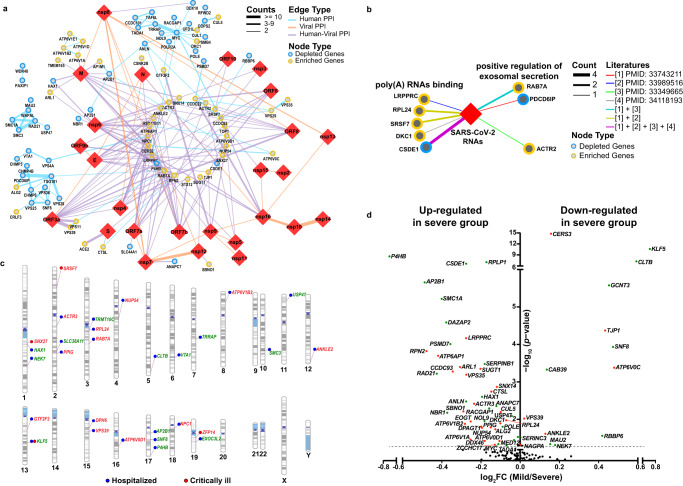
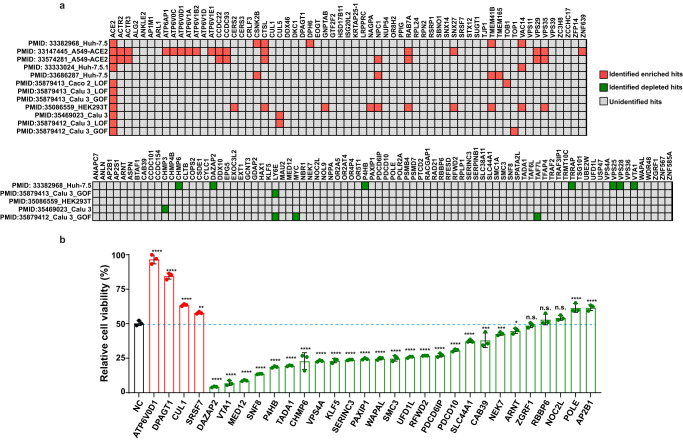
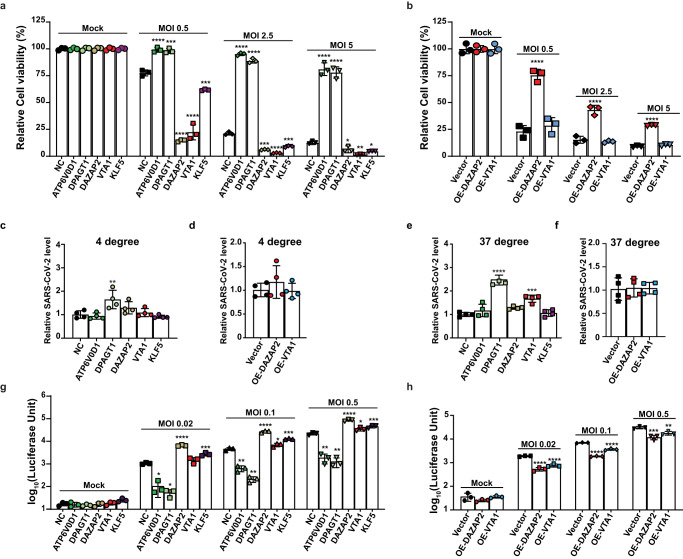
Host anti-viral factors are essential for controlling SARS-CoV-2 infection but remain largely unknown due to the biases of previous large-scale studies toward pro-viral host factors. To fill in this knowledge gap, we perform a genome-wide CRISPR dropout screen and integrate analyses of the multi-omics data of the CRISPR screen, genome-wide association studies, single-cell RNA-Seq, and host-virus proteins or protein/RNA interactome. This study uncovers many host factors that are currently underappreciated, including the components of V-ATPases, ESCRT, and N-glycosylation pathways that modulate viral entry and/or replication. The cohesin complex is also identified as an anti-viral pathway, suggesting an important role of three-dimensional chromatin organization in mediating host-viral interaction. Furthermore, we discover another anti-viral regulator KLF5, a transcriptional factor involved in sphingolipid metabolism, which is up-regulated, and harbors genetic variations linked to COVID-19 patients with severe symptoms. Anti-viral effects of three identified candidates (DAZAP2/VTA1/KLF5) are confirmed individually. Molecular characterization of DAZAP2/VTA1/KLF5-knockout cells highlights the involvement of genes related to the coagulation system in determining the severity of COVID-19. Together, our results provide further resources for understanding the host anti-viral network during SARS-CoV-2 infection and may help develop new countermeasure strategies.
© 2024. The Author(s).
Conflict of interest statement
W.P. served as an advisor for Fresh wind biotechnologies. X.X. and P.-Y.S. have filed a patent on the reverse genetic system and reporter SARS-CoV-2. The remaining authors declare no competing interests.
Figures
Update of
-
Integrated multi-omics analyses identify key anti-viral host factors and pathways controlling SARS-CoV-2 infection.Res Sq [Preprint]. 2022 Aug 15:rs.3.rs-1910932. doi: 10.21203/rs.3.rs-1910932/v1. Res Sq. 2022. Update in: Nat Commun. 2024 Jan 2;15(1):109. doi: 10.1038/s41467-023-44175-1. PMID: 36032971 Free PMC article. Updated. Preprint.
References
-
- Johns Hopkins University Coronavirus Resource Center. Mortality analyses. https://coronavirus.jhu.edu/data/mortality (2023).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
