

Inference of Infectious Disease Transmission through a Relaxed Bottleneck Using Multiple Genomes Per Host
- PMID: 38168711
- PMCID: PMC10798190
- DOI: 10.1093/molbev/msad288
Inference of Infectious Disease Transmission through a Relaxed Bottleneck Using Multiple Genomes Per Host
Abstract
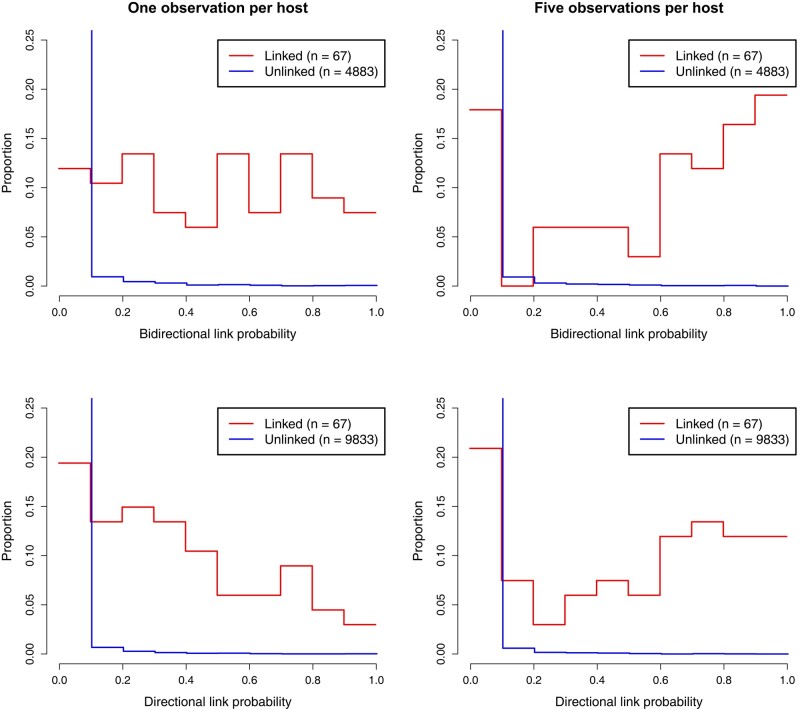
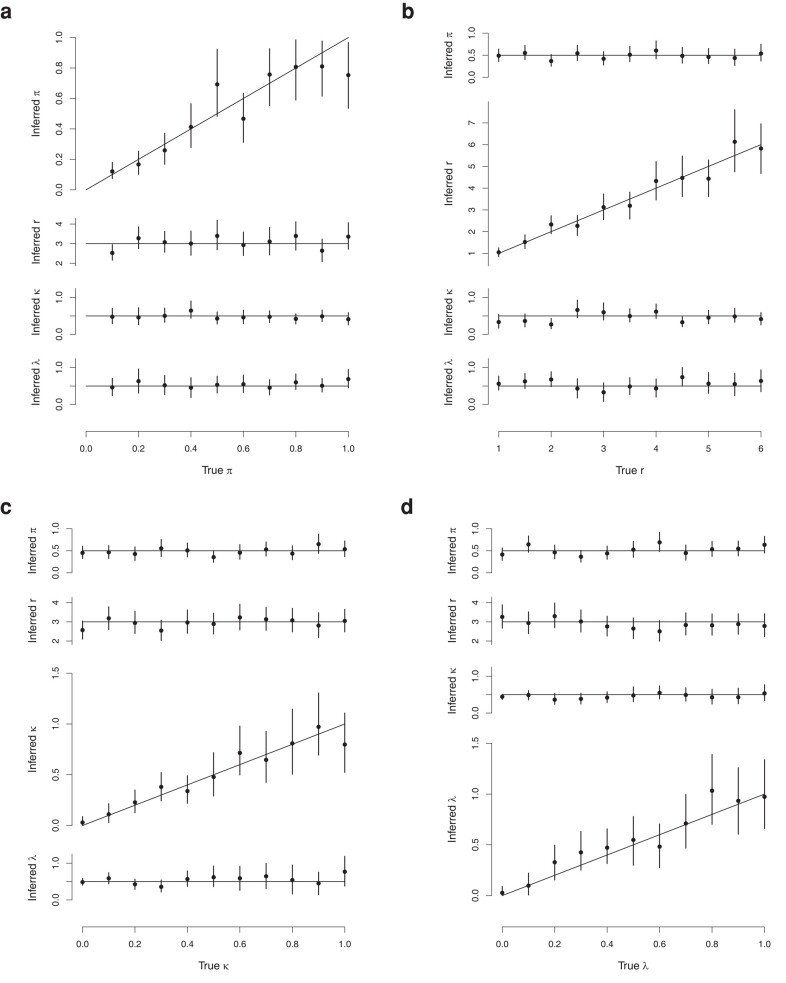
In recent times, pathogen genome sequencing has become increasingly used to investigate infectious disease outbreaks. When genomic data is sampled densely enough amongst infected individuals, it can help resolve who infected whom. However, transmission analysis cannot rely solely on a phylogeny of the genomes but must account for the within-host evolution of the pathogen, which blurs the relationship between phylogenetic and transmission trees. When only a single genome is sampled for each host, the uncertainty about who infected whom can be quite high. Consequently, transmission analysis based on multiple genomes of the same pathogen per host has a clear potential for delivering more precise results, even though it is more laborious to achieve. Here, we present a new methodology that can use any number of genomes sampled from a set of individuals to reconstruct their transmission network. Furthermore, we remove the need for the assumption of a complete transmission bottleneck. We use simulated data to show that our method becomes more accurate as more genomes per host are provided, and that it can infer key infectious disease parameters such as the size of the transmission bottleneck, within-host growth rate, basic reproduction number, and sampling fraction. We demonstrate the usefulness of our method in applications to real datasets from an outbreak of Pseudomonas aeruginosa amongst cystic fibrosis patients and a nosocomial outbreak of Klebsiella pneumoniae.
Keywords: genomic epidemiology; infectious disease outbreak; transmission analysis; within-host diversity and evolution.
© The Author(s) 2024. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures




Similar articles
-
Within-host diversity improves phylogenetic and transmission reconstruction of SARS-CoV-2 outbreaks.Elife. 2023 Sep 21;12:e84384. doi: 10.7554/eLife.84384. Elife. 2023. PMID: 37732733 Free PMC article.
-
Genomic Infectious Disease Epidemiology in Partially Sampled and Ongoing Outbreaks.Mol Biol Evol. 2017 Apr 1;34(4):997-1007. doi: 10.1093/molbev/msw275. Mol Biol Evol. 2017. PMID: 28100788 Free PMC article.
-
When are pathogen genome sequences informative of transmission events?PLoS Pathog. 2018 Feb 8;14(2):e1006885. doi: 10.1371/journal.ppat.1006885. eCollection 2018 Feb. PLoS Pathog. 2018. PMID: 29420641 Free PMC article.
-
The role of pathogen genomics in assessing disease transmission.BMJ. 2015 May 11;350:h1314. doi: 10.1136/bmj.h1314. BMJ. 2015. PMID: 25964672 Review.
-
Progress and challenges in virus genomic epidemiology.Trends Parasitol. 2021 Dec;37(12):1038-1049. doi: 10.1016/j.pt.2021.08.007. Epub 2021 Oct 4. Trends Parasitol. 2021. PMID: 34620561 Review.
Cited by
-
A graph homomorphism approach for unraveling histories of metastatic cancers and viral outbreaks under evolutionary constraints.Nat Commun. 2025 Aug 28;16(1):8027. doi: 10.1038/s41467-025-63411-4. Nat Commun. 2025. PMID: 40877260 Free PMC article.
-
A Bayesian modelling framework with model comparison for epidemics with super-spreading.Infect Dis Model. 2025 Aug 5;10(4):1418-1432. doi: 10.1016/j.idm.2025.07.017. eCollection 2025 Dec. Infect Dis Model. 2025. PMID: 40822275 Free PMC article.
-
Incorporating Epidemiological Data into the Genomic Analysis of Partially Sampled Infectious Disease Outbreaks.Mol Biol Evol. 2025 Apr 1;42(4):msaf083. doi: 10.1093/molbev/msaf083. Mol Biol Evol. 2025. PMID: 40256930 Free PMC article.
References
-
- Brooks SP, Gelman A. General methods for monitoring convergence of iterative simulations. J Comput Graph Stat. 1998:7:434–455.
-
- Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Reacher M, Haworth CS, Curran MD, Harris SR, et al. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet. 2013:381:1551–1560. 10.1016/S0140-6736(13)60632-7. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
