

doi: 10.1038/s41587-023-02019-9.
Epub 2024 Jan 2.
Inferring super-resolution tissue architecture by integrating spatial transcriptomics with histology
Affiliations
- PMID: 38168986
- PMCID: PMC11260191
- DOI: 10.1038/s41587-023-02019-9
Item in Clipboard
Inferring super-resolution tissue architecture by integrating spatial transcriptomics with histology
Nat Biotechnol.
2024 Sep.
Abstract
Spatial transcriptomics (ST) has demonstrated enormous potential for generating intricate molecular maps of cells within tissues. Here we present iStar, a method based on hierarchical image feature extraction that integrates ST data and high-resolution histology images to predict spatial gene expression with super-resolution. Our method enhances gene expression resolution to near-single-cell levels in ST and enables gene expression prediction in tissue sections where only histology images are available.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures

Comparison of iStar’s super-resolution gene expression patterns with the paired histology image in the Xenium-derived pseudo-Visium data. Spot boundaries are highlighted.

Visualization of predicted super-resolution gene expressions by iStar at various scales of resolution enhancement for three breast cancer-related genes (ESR1, ERBB2, and PGR) in the Xenium-derived pseudo-Visium data.

Prediction accuracy of iStar and XFuse as measured by root mean squared error (RMSE) and structural similarity index measure (SSIM) for all the 313 genes in the Xenium-derived pseudo-Visium data obtained from a breast cancer patient. In each scatter plot, a dot represents a gene. In this analysis, Section 1 was first treated as the training sample in which in-sample prediction was perfromed for Section 1 and then out-of-sample prediction was perfromed for Section 2. We then repeated this analysis by treating Section 2 as the ‘in-sample’ and Section 1 as the ‘out-of-sample’. The evaluation metrics in’Average’ is the average of those in ‘Sections 1’ and ‘Section 2’.

Visualization of single-cell level gene expression predicted by iStar for the pseudo-Visium breast cancer data derived from Xenium data. Shown on the left is the ground truth single-cell level gene expression directly measured by Xenium, and shown on the right is the single-cell level gene expression predicted by iStar. For each gene, the root mean squared error (RMSE) and Pearson’s correlation coefficient (PCC) between the prediction and the ground truth across the whole tissue and within the shown region are displayed.

Prediction accuracy of iStar and XFuse for single-cell level gene expression prediction as measured by root mean squared error (RMSE) for all the 313 genes in the Xenium-derived pseudo-Visium data obtained from a breast cancer patient. In each scatter plot, a dot represents a gene. In this analysis, Section 1 was first treated as the training sample in which in-sample prediction was performed for Section 1 and then out-of-sample prediction was performed for Section 2. We then repeated this analysis by treating Section 2 as the ‘in-sample’ and Section 1 as the ‘out-of-sample’. The evaluation metrics in’Average’ is the average of those in ‘Sections 1’ and ‘Section 2’. We stratified cells by the quantiles of their cell size.

iStar detected tertiary lymphoid structures (TLSs) in the Anderson et al. (2021) HER2+ breast cancer dataset. Displayed are the predicted TLS scores by iStar and the original publication, along with the pathologist’s manual annotation reported in the original publication. Super-resolution was performed with 128x resolution enhancement.

Visualization of the spot-level training data, ground truth gene expression, and predicted super-resolution gene expressions by iStar and XFuse for 24 highly variable genes, whose variances are in the 80%−100% quantiles among all the 248 genes in the Xenium-derived pseudo-Visium data obtained from mouse brain. The variance quantiles of the genes in the order of top-left, top-right, bottom-left, bottom-right are equally spaced from 100% to 80% (in descending order). Super-resolution gene expressions are visualized at the scale of 8x resolution enhancement.

a. Prediction accuracy of by iStar and XFuse as measured by root mean squared error (RMSE) and structural similarity index measure (SSIM) for all the 248 genes in the Xenium-derived pseudo-Visium data obtained from mouse brain. In each scatter plot, a dot represents one gene. b. Segmentation of the Xenium-derived pseudo-Visium data obtained from mouse brain by iStar and XFuse using all 248 genes available in this dataset. Super-resolution was performed with 128x resolution enhancement.

Analyses of a. mouse brain (coronal cut), b. mouse brain posterior (sagittal cut), and c. mouse brain ofactory bulb Visium datasets generated by 10x Genomics. For each dataset, iStar was applied to enhance the resolution of the top 1000 most highly variable genes. Super-resolution was performed with 128x resolution enhancement. Segmentations by iStar identified fine-grained tissue structures in the mouse brain and agreed with the Allen Brain Atlas annotations.

Comparison of gene-based segmentation by iStar with manual tissue annotation in a. mouse kidney and b. prostate cancer Visium datasets generated by 10x Genomics. c. Gene-based segmentation by iStar in a colorectal cancer Visium dataset by 10x Genomics. d. Comparison of tertiary lymphoid structure (TLS) signature score by iStar with manual TLS annotation in a kidney cancer Visium dataset generated by Meylan et al. (2022).
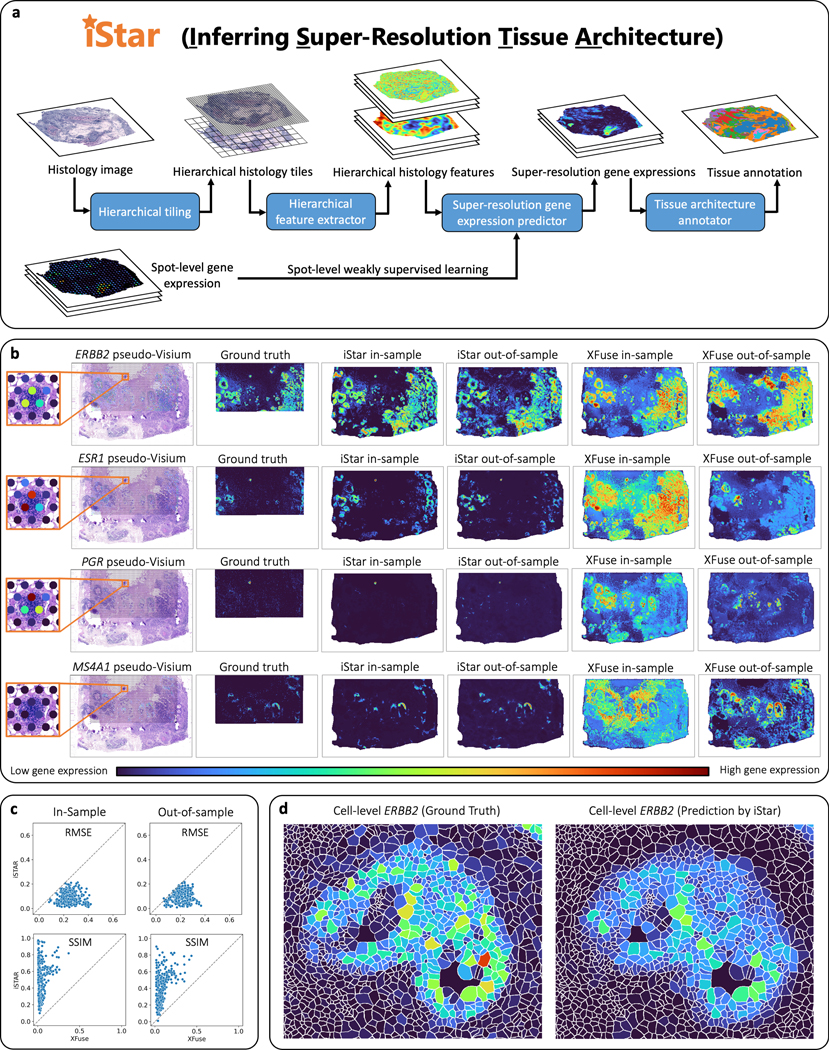
a, Model summary of iStar. The histology image is hierarchically divided into tiles, which are then converted into hierarchical histology image features. These features, combined with the spot-level gene expression data, are then utilized to predict super-resolution gene expression. Finally, tissue architecture is inferred based on the super-resolved gene expression prediction. b, c, Evaluation of super-resolution gene expression prediction accuracy using the Xenium breast cancer dataset, which includes two consecutively cut tissue sections. The Xenium data served as the ground truth and were used to simulate spot-level gene expression based on the spot size and layout of Visium. For in-sample prediction, both model training and prediction were performed using section 2’s pseudo-Visium data. For out-of-sample prediction, section 1 was used for model training and section 2 was used for prediction in which only its histology image was used as input. b, Visual comparison between iStar and XFuse. ERBB2, ESR1, and PGR are genes that encode biomarkers for breast cancer prognosis, while MS4A1 is a B cell marker gene. Super-resolution gene expressions are visualized at the scale of 8x resolution enhancement. Visualizations of additional genes at the scale of 8x resolution enhancement are presented in Supplementary Figs. 1–5. Visualizations at other resolutions are available in Supplementary Fig. 6 and at https://upenn.box.com/v/istar-results-benchmark . c, Numerical comparison between iStar and XFuse for 128x resolution enhancement of gene expression. The degree of resolution enhancement is defined as the number of superpixels in the super-resolution prediction divided by the number of spots in the training data. Each dot represents one of the 313 genes. Additional numerical evaluations are reported in Supplementary Fig. 7. d, Predicted single cell-level gene expression, which was computed from the predicted superpixel-level gene expression using the cell segmentation masks provided in the dataset. Additional examples are presented in Supplementary Fig. 9.

a, Comparison of unsupervised tissue segmentation by iStar and XFuse with manual annotation of one of the two consecutively cut tissue sections of a breast cancer patient in the Xenium dataset. The model was trained using the pseudo-Visium spot-level gene expression simulated from Section 1 (in-sample) of the Xenium data. Section 2 was treated as the out-of-sample section, and its super-resolution gene expression was predicted only using its histology image. Super-resolution was performed with 128x resolution enhancement. b, Tissue architecture annotation of a breast cancer tissue in the HER2ST breast cancer dataset (three consecutively cut tissue sections in Subject H). Super-resolution was performed with 128x resolution enhancement. c, iStar assigned biologically meaningful labels to the tissue clusters by performing superpixel-level cell type inference, followed by a cell type enrichment analysis, where depletion, i.e., negative enrichment, was not shown in the heatmap. d, iStar’s unsupervised tissue segmentation revealed intraltumoral heterogeneity that agreed with the pathologist’s manual annotation. e, A small cancer region detected by iStar that was missed in manual annotation provided in the original publication. f, Detection of Tertiary Lymphoid Structures (TLS) in the HER2ST breast cancer dataset (Subject G) by iStar. The TLS score was calculated as the mean of the standardized super-resolution gene expressions of the TLS marker genes shown in Supplementary Table 1.
References
-
- Burgess DJ Spatial transcriptomics coming of age. Nat. Rev. Genet 20, 317 (2019). - PubMed
-
- Asp M, Bergenstrahle J. & Lundeberg J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. Bioessays 42, e1900221 (2020). - PubMed
-
- Crosetto N, Bienko M. & van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat. Rev. Genet 16, 57–66 (2015). - PubMed
-
- Moor AE & Itzkovitz S. Spatial transcriptomics: paving the way for tissue-level systems biology. Curr. Opin. Biotechnol 46, 126–133 (2017). - PubMed
-
- Hu J. et al. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 18, 1342–1351 (2021). - PubMed
Methods-only References
-
- Steiner A. et al. How to train your vit? data, augmentation, and regularization in vision transformers. Preprint at arXiv, 10.48550/arXiv.2106.10270 (2021). - DOI
-
- Xu B, Wang N, Chen T. & Li M. Empirical evaluation of rectified activations in convolutional network. Preprint arXiv, 10.48550/arXiv.1505.00853 (2015). - DOI
-
- Clevert D-A, Unterthiner T. & Hochreiter S. Fast and accurate deep network learning by exponential linear units (ELUs). Preprint at arXiv, 10.48550/arXiv.1511.07289 (2015). - DOI
MeSH terms
Grants and funding
- R01EY030192/U.S. Department of Health & Human Services | NIH | National Eye Institute (NEI)
- P01AG066597/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
- R01GM125301/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- R01 GM125301/GM/NIGMS NIH HHS/United States
- R01CA266280/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P30 CA082709/CA/NCI NIH HHS/United States
- U01CA264583/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P01 AG066597/AG/NIA NIH HHS/United States
- R01 CA266280/CA/NCI NIH HHS/United States
- R01 HG013185/HG/NHGRI NIH HHS/United States
- UL1 TR001873/TR/NCATS NIH HHS/United States
- R01 EY030192/EY/NEI NIH HHS/United States
- R01HG013158/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- R01 HL150359/HL/NHLBI NIH HHS/United States
- R01HL150359/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
LinkOut - more resources
Full Text Sources
