

Disruption of MAM integrity in mutant FUS oligodendroglial progenitors from hiPSCs
- PMID: 38170217
- PMCID: PMC10764485
- DOI: 10.1007/s00401-023-02666-x
Disruption of MAM integrity in mutant FUS oligodendroglial progenitors from hiPSCs
Erratum in
-
Correction to: Disruption of MAM integrity in mutant FUS oligodendroglial progenitors from hiPSCs.Acta Neuropathol. 2024 Jun 13;147(1):99. doi: 10.1007/s00401-024-02748-4. Acta Neuropathol. 2024. PMID: 38871840 Free PMC article. No abstract available.
Abstract
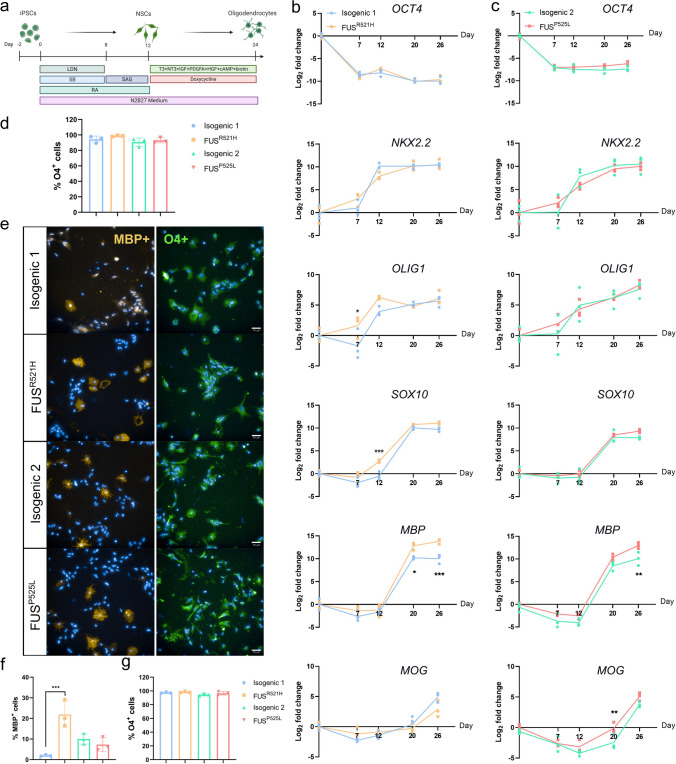
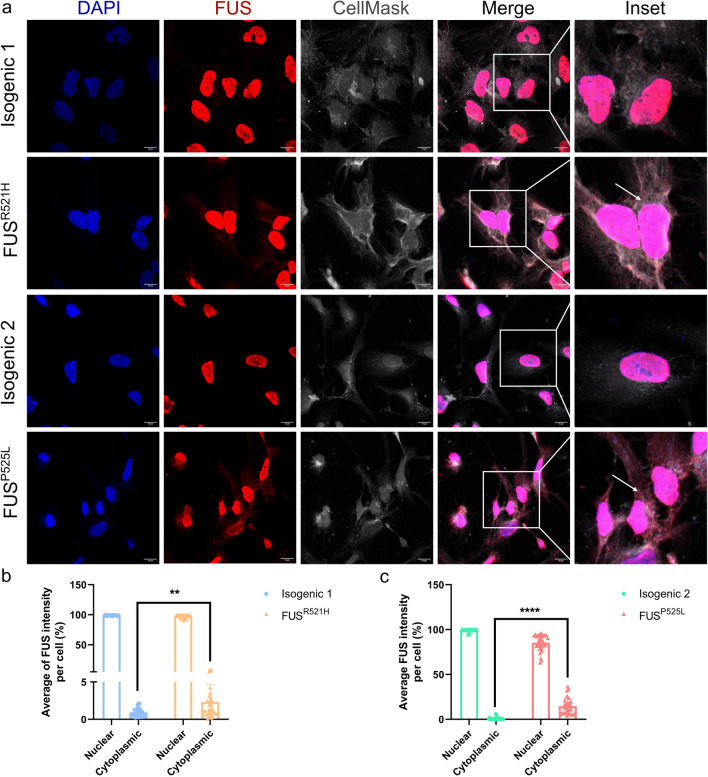
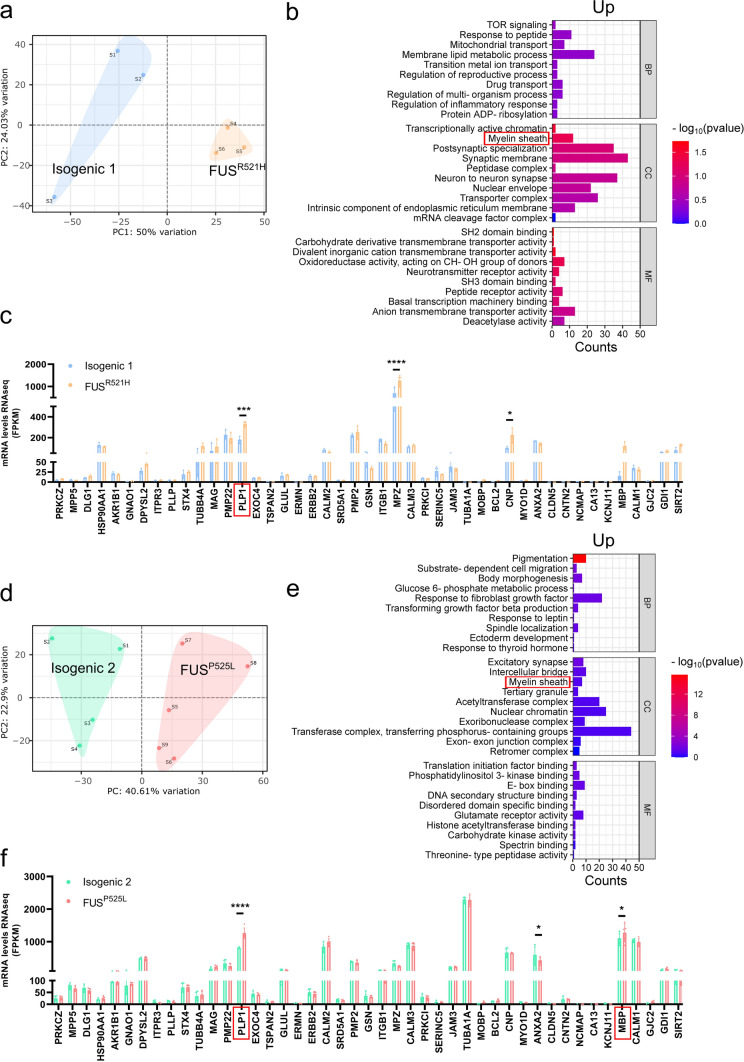
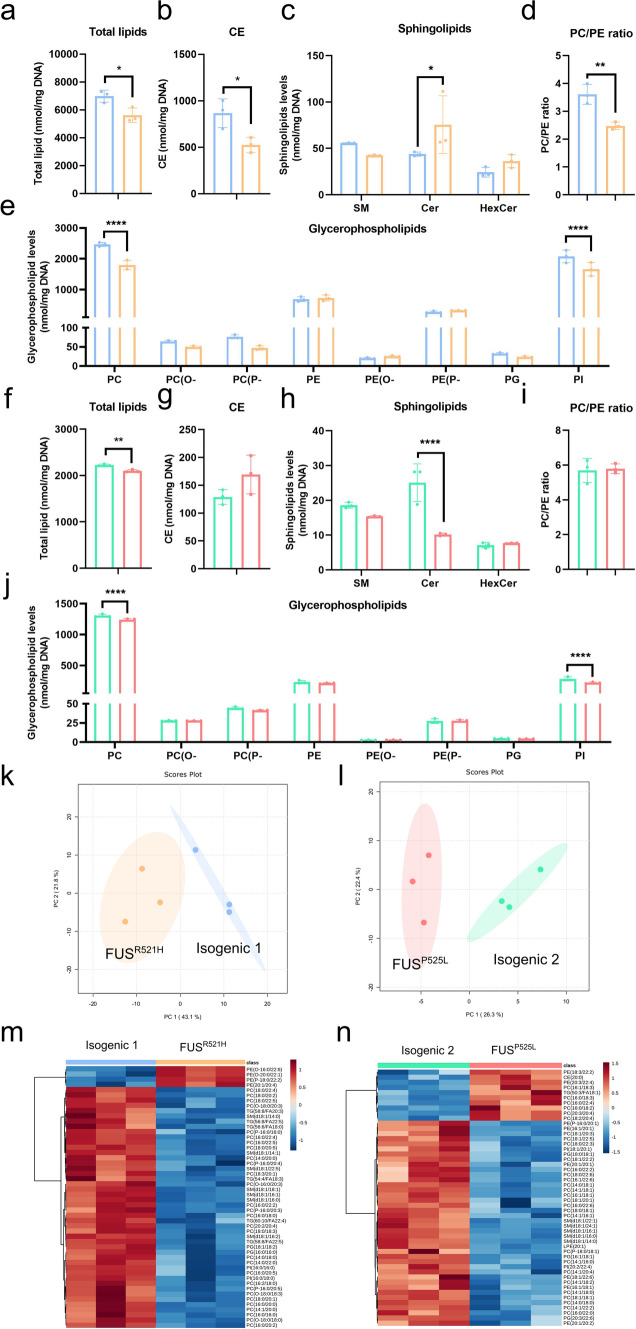
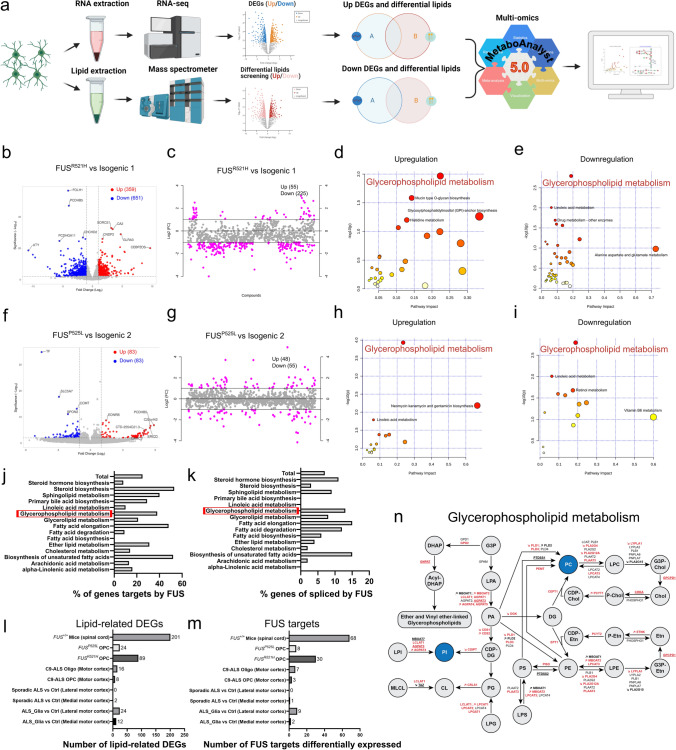
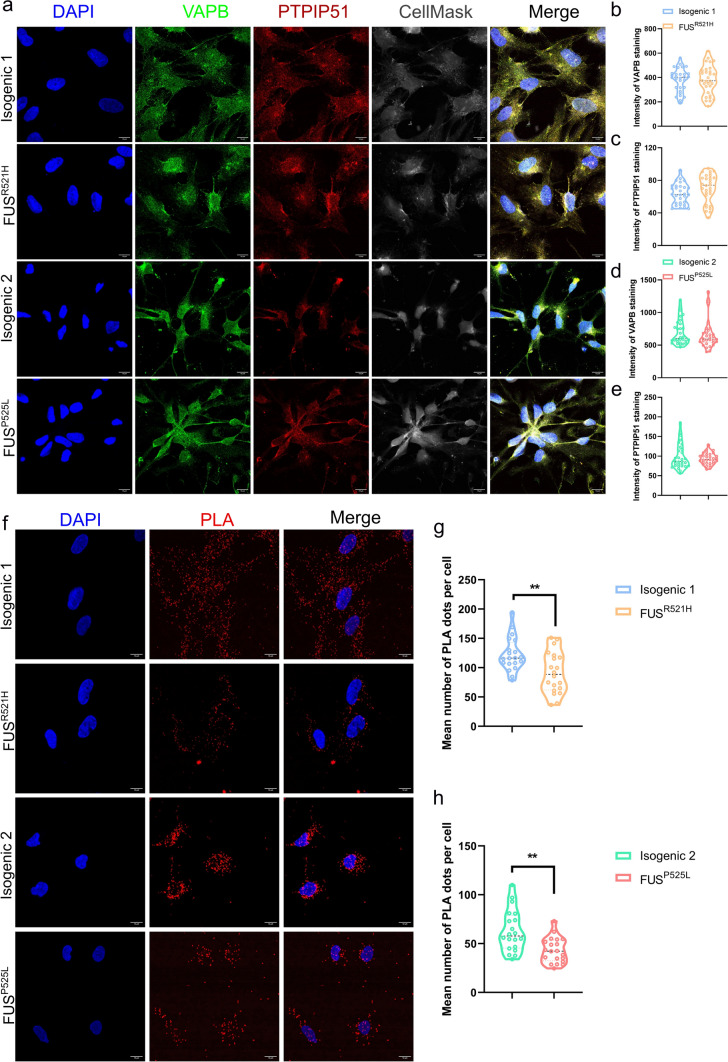
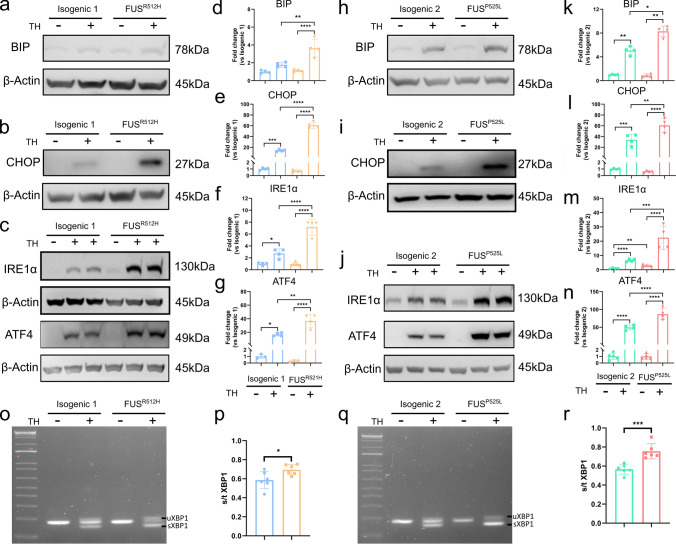
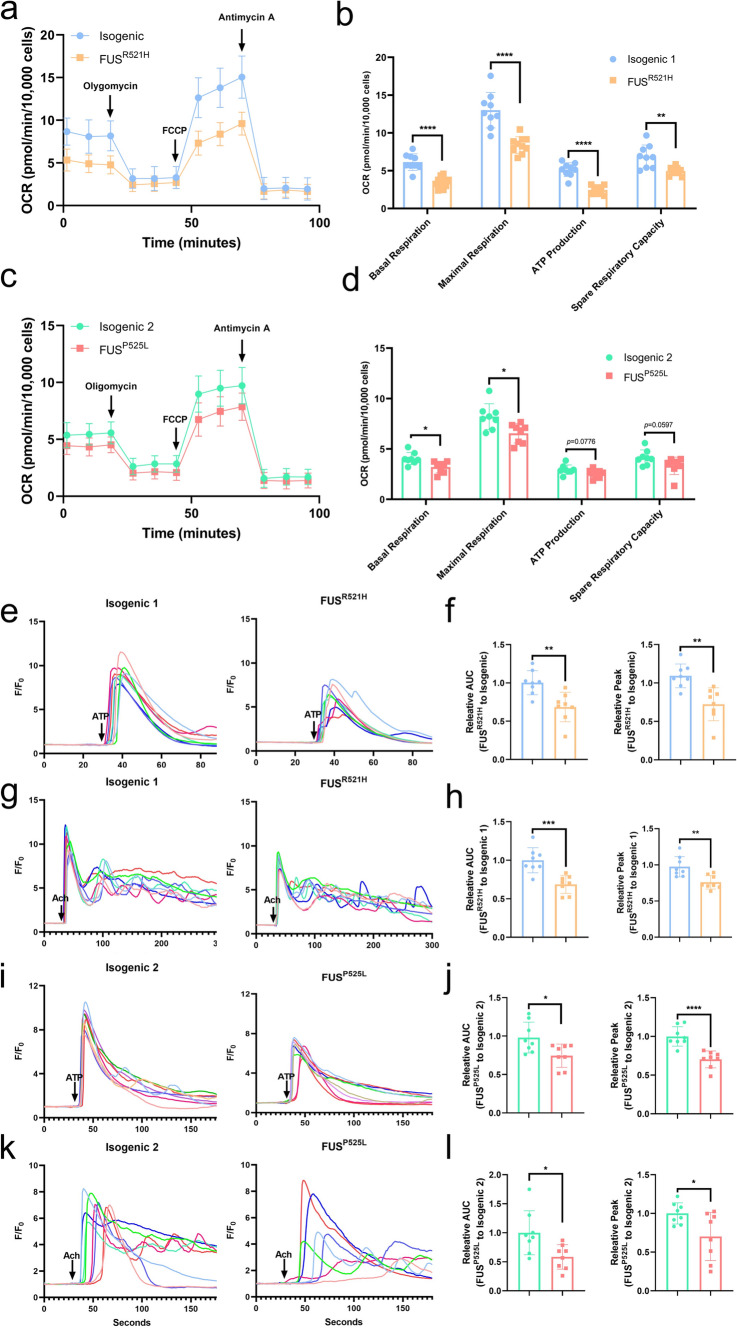
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive and fatal neurodegenerative disorder, characterized by selective loss of motor neurons (MNs). A number of causative genetic mutations underlie the disease, including mutations in the fused in sarcoma (FUS) gene, which can lead to both juvenile and late-onset ALS. Although ALS results from MN death, there is evidence that dysfunctional glial cells, including oligodendroglia, contribute to neurodegeneration. Here, we used human induced pluripotent stem cells (hiPSCs) with a R521H or a P525L mutation in FUS and their isogenic controls to generate oligodendrocyte progenitor cells (OPCs) by inducing SOX10 expression from a TET-On SOX10 cassette. Mutant and control iPSCs differentiated efficiently into OPCs. RNA sequencing identified a myelin sheath-related phenotype in mutant OPCs. Lipidomic studies demonstrated defects in myelin-related lipids, with a reduction of glycerophospholipids in mutant OPCs. Interestingly, FUSR521H OPCs displayed a decrease in the phosphatidylcholine/phosphatidylethanolamine ratio, known to be associated with maintaining membrane integrity. A proximity ligation assay further indicated that mitochondria-associated endoplasmic reticulum membranes (MAM) were diminished in both mutant FUS OPCs. Moreover, both mutant FUS OPCs displayed increased susceptibility to ER stress when exposed to thapsigargin, and exhibited impaired mitochondrial respiration and reduced Ca2+ signaling from ER Ca2+ stores. Taken together, these results demonstrate a pathological role of mutant FUS in OPCs, causing defects in lipid metabolism associated with MAM disruption manifested by impaired mitochondrial metabolism with increased susceptibility to ER stress and with suppressed physiological Ca2+ signaling. As such, further exploration of the role of oligodendrocyte dysfunction in the demise of MNs is crucial and will provide new insights into the complex cellular mechanisms underlying ALS.
Keywords: Amyotrophic lateral sclerosis; ER stress; FUS; Lipid defects; MAM disruption; Mitochondrial dysfunction.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
-
- Angebault C, Fauconnier J, Patergnani S, Rieusset J, Danese A, Affortit CA, Jagodzinska J, Mégy C, Quiles M, Cazevieille C, Korchagina J, Bonnet-Wersinger D, Milea D, Hamel C, Pinton P, Thiry M, Lacampagne A, Delprat B, Delettre C. ER-mitochondria cross-talk is regulated by the Ca(2+) sensor NCS1 and is impaired in Wolfram syndrome. Sci Signal. 2018 doi: 10.1126/scisignal.aaq1380. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 12ZG121N/Fonds Wetenschappelijk Onderzoek
- SBO-S001221N/Fonds Wetenschappelijk Onderzoek
- 12AIK24N/Fonds Wetenschappelijk Onderzoek
- W001422N/Fonds Wetenschappelijk Onderzoek
- W001422N/Fonds Wetenschappelijk Onderzoek
- 201908440360/China Scholarship Council
- C14-17-107/Universitaire Ziekenhuizen Leuven, KU Leuven
- C14/22/132/Universitaire Ziekenhuizen Leuven, KU Leuven
- ALS-OL/Fondation Thierry Latran
- C14/19/099/Research Council of the KU Leuven
- AKUL/19/34/Research Council of the KU Leuven
- iBOF/23/02/the ALS Liga ('A Cure for ALS')
- IDN/22/012/Generet Award for Rare Diseases
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
