

Splicing modulators impair DNA damage response and induce killing of cohesin-mutant MDS and AML
- PMID: 38170787
- PMCID: PMC11222919
- DOI: 10.1126/scitranslmed.ade2774
Splicing modulators impair DNA damage response and induce killing of cohesin-mutant MDS and AML
Abstract
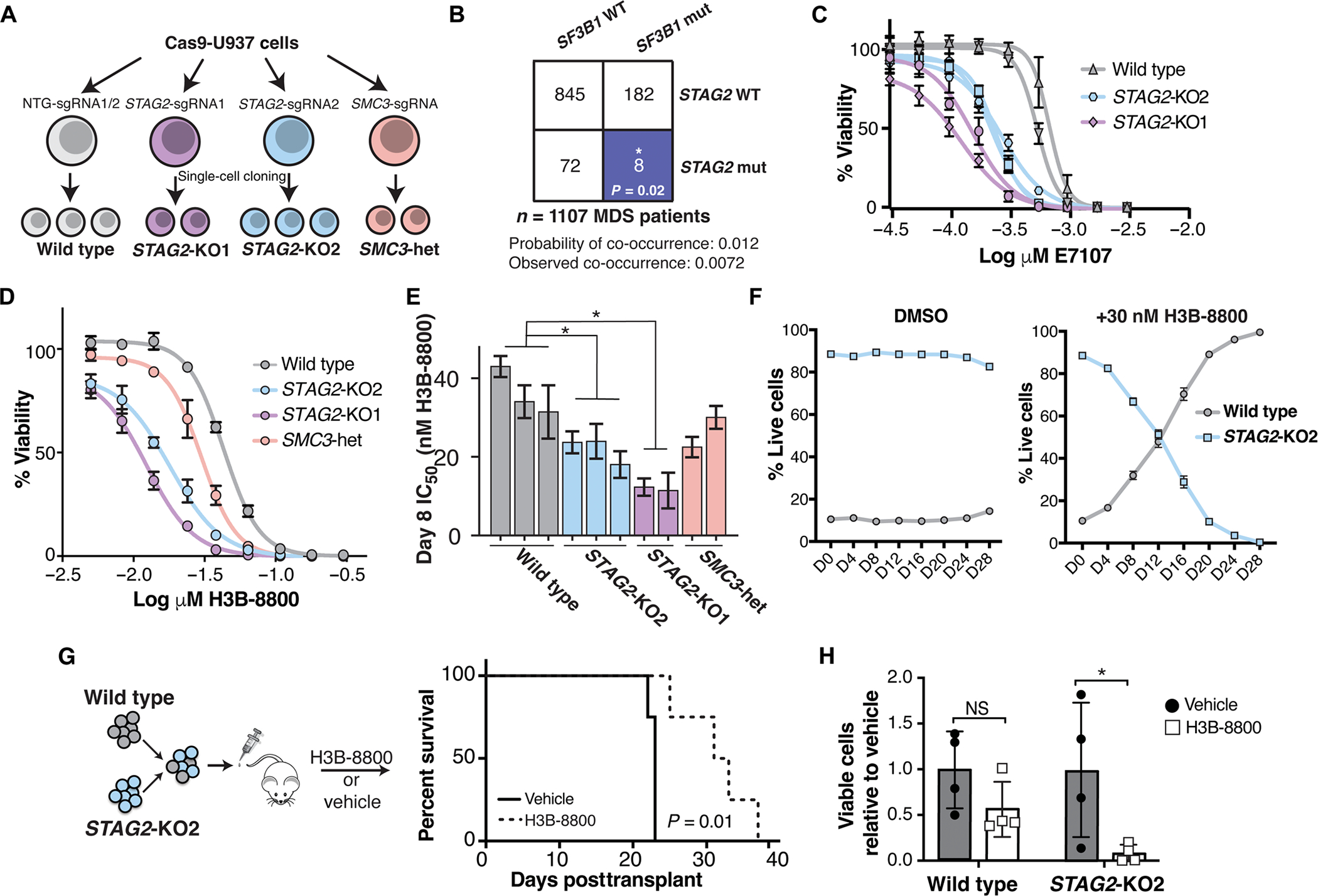
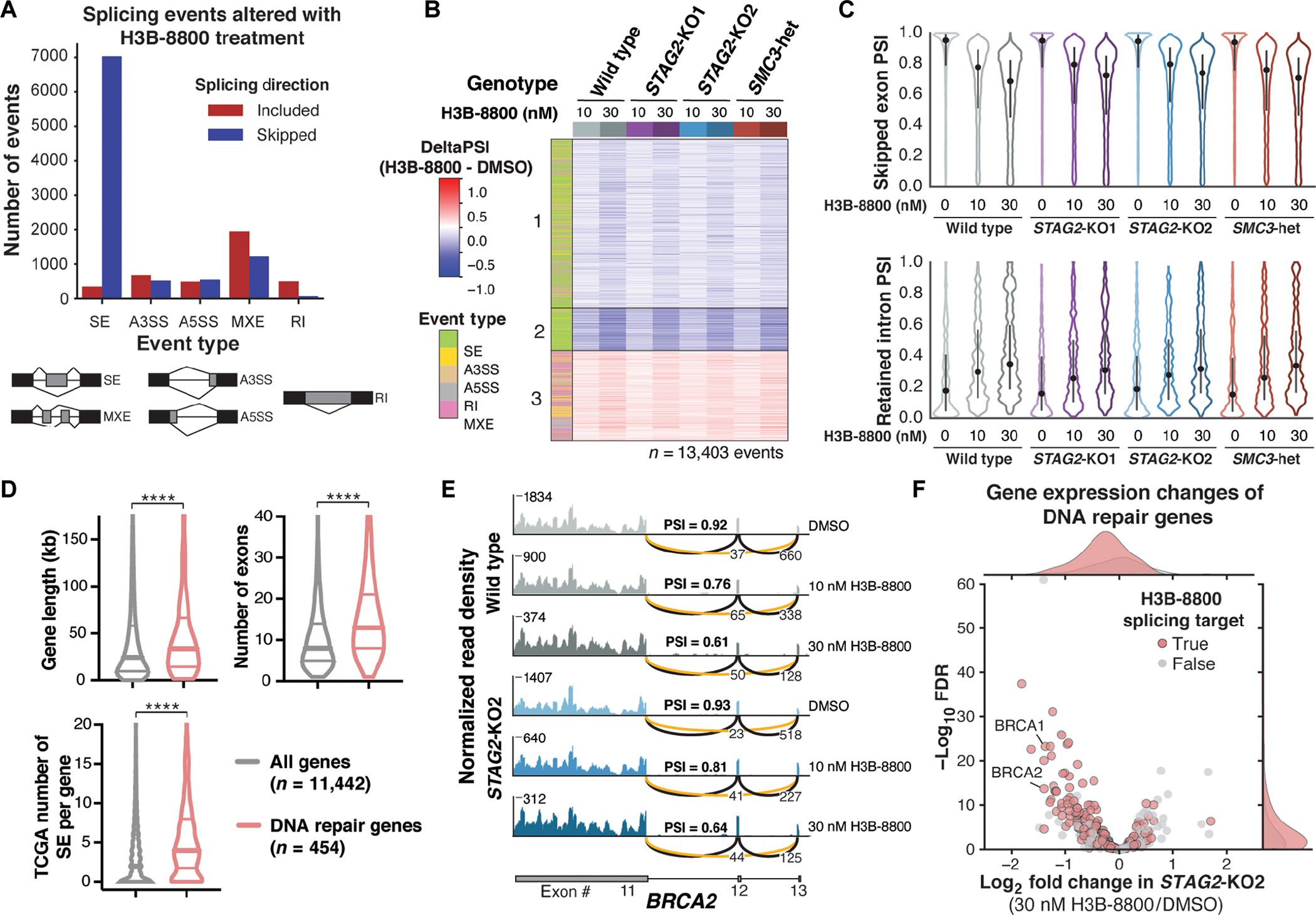
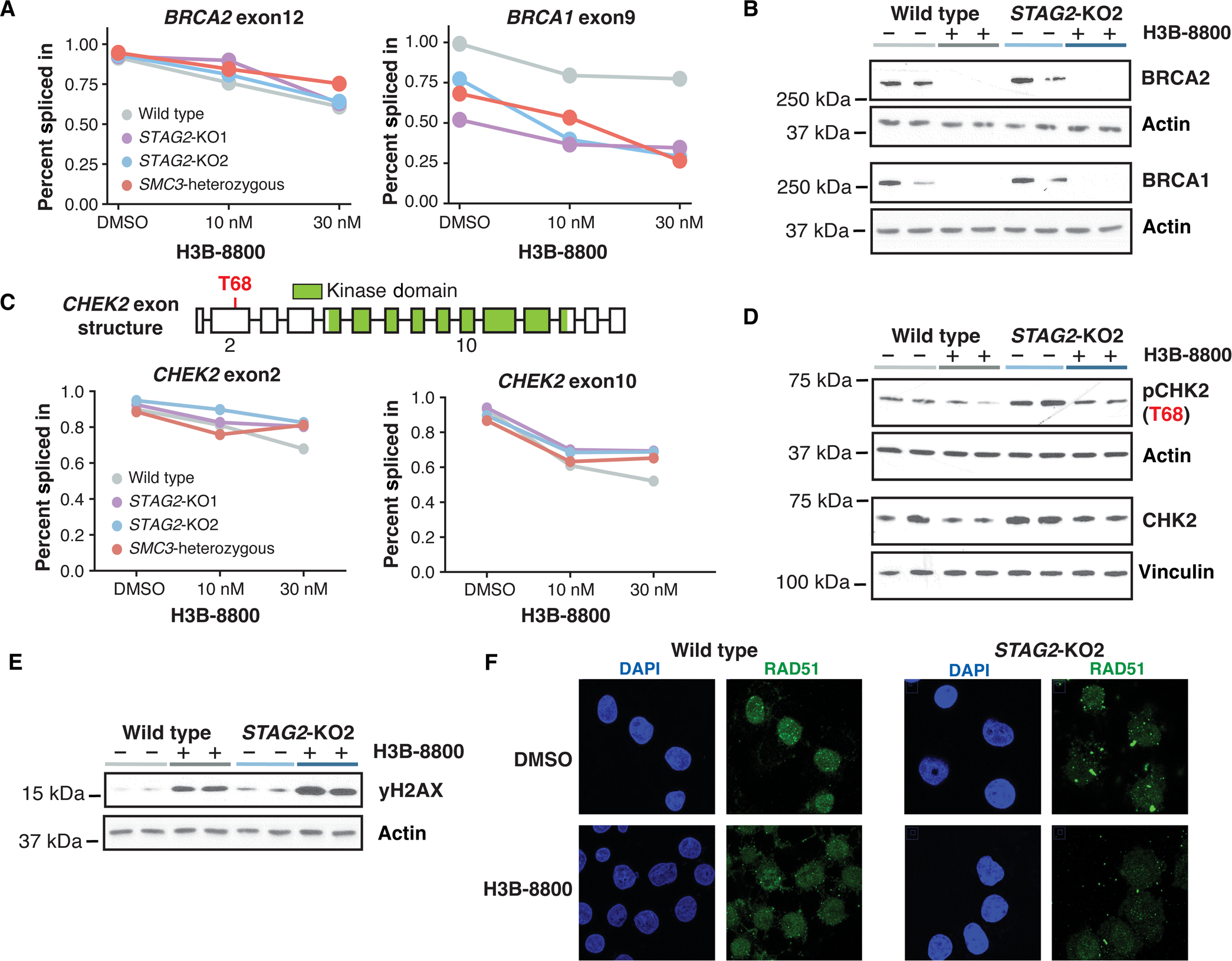
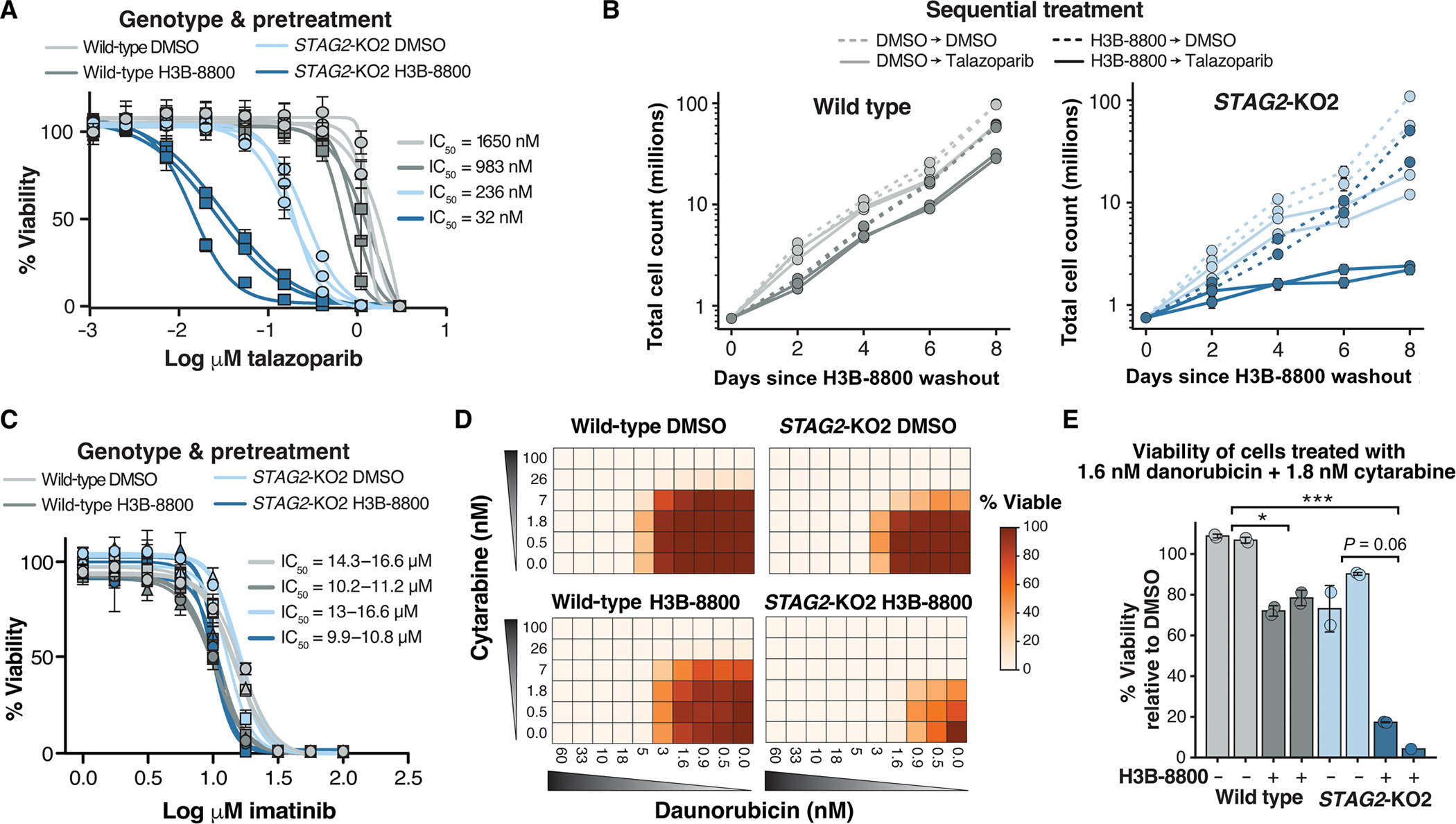
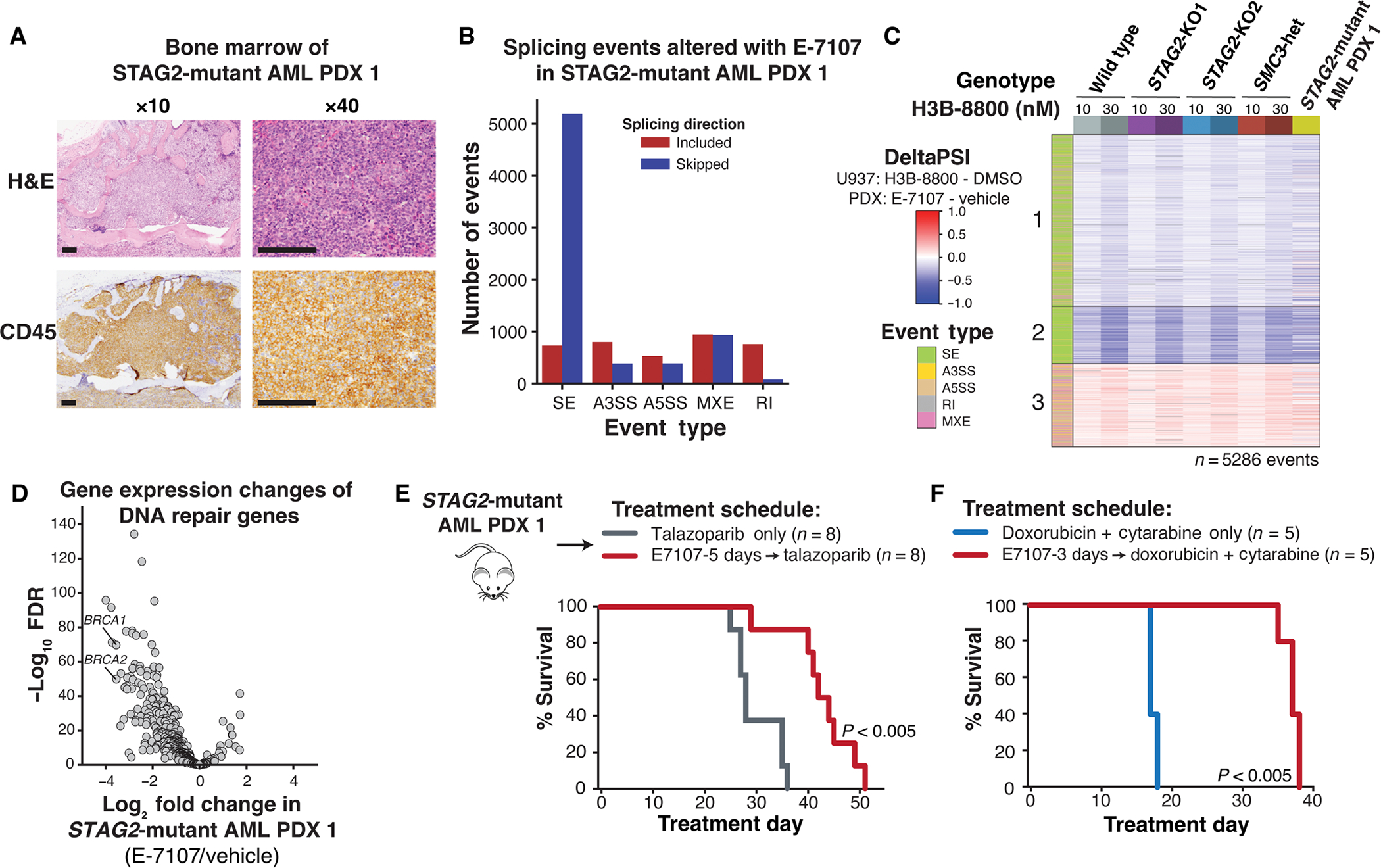
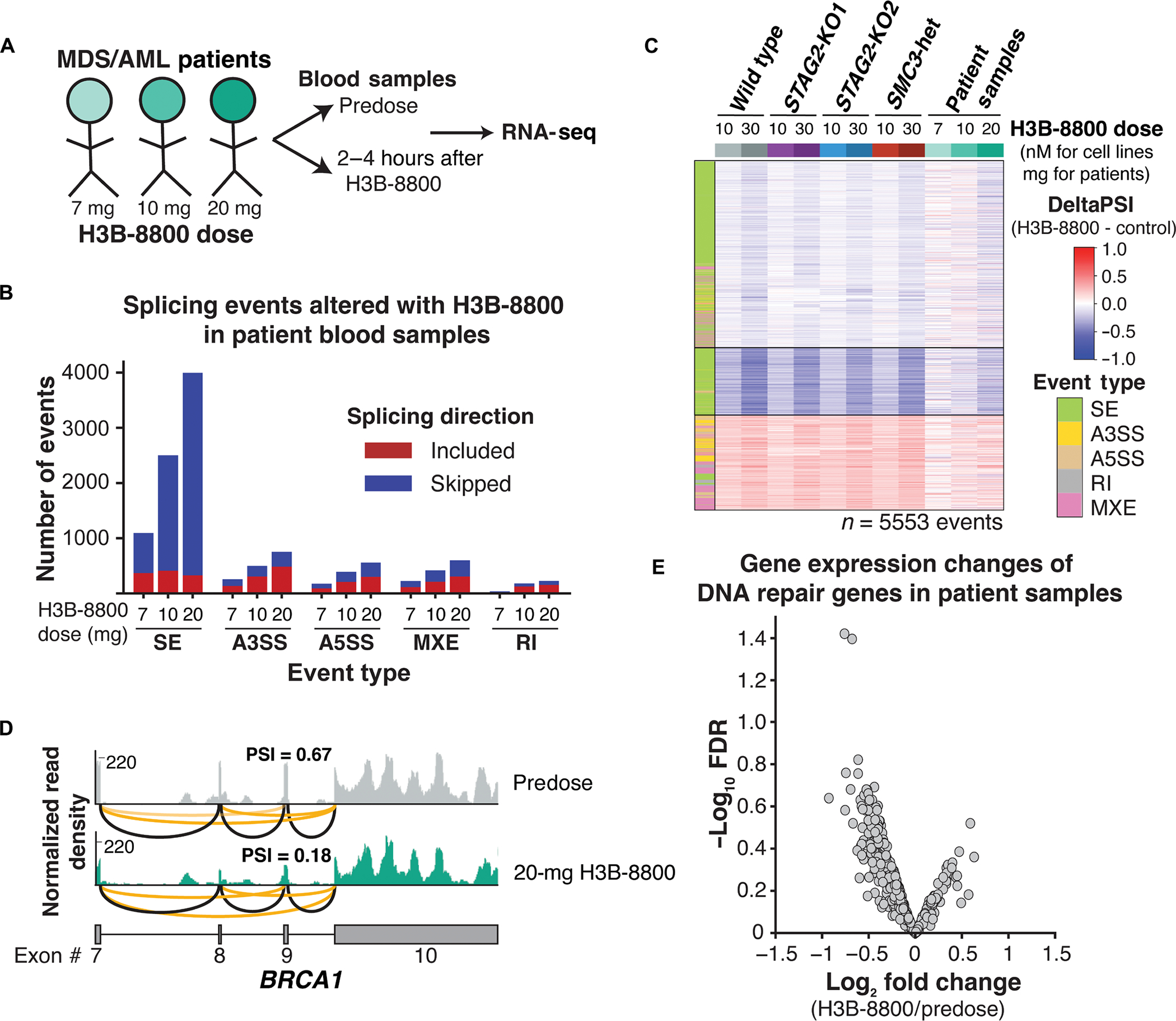
Splicing modulation is a promising treatment strategy pursued to date only in splicing factor-mutant cancers; however, its therapeutic potential is poorly understood outside of this context. Like splicing factors, genes encoding components of the cohesin complex are frequently mutated in cancer, including myelodysplastic syndromes (MDS) and secondary acute myeloid leukemia (AML), where they are associated with poor outcomes. Here, we showed that cohesin mutations are biomarkers of sensitivity to drugs targeting the splicing factor 3B subunit 1 (SF3B1) H3B-8800 and E-7107. We identified drug-induced alterations in splicing, and corresponding reduced gene expression, of a number of DNA repair genes, including BRCA1 and BRCA2, as the mechanism underlying this sensitivity in cell line models, primary patient samples and patient-derived xenograft (PDX) models of AML. We found that DNA damage repair genes are particularly sensitive to exon skipping induced by SF3B1 modulators due to their long length and large number of exons per transcript. Furthermore, we demonstrated that treatment of cohesin-mutant cells with SF3B1 modulators not only resulted in impaired DNA damage response and accumulation of DNA damage, but it sensitized cells to subsequent killing by poly(ADP-ribose) polymerase (PARP) inhibitors and chemotherapy and led to improved overall survival of PDX models of cohesin-mutant AML in vivo. Our findings expand the potential therapeutic benefits of SF3B1 splicing modulators to include cohesin-mutant MDS and AML.
Figures
References
-
- Losada A, Cohesin in cancer: Chromosome segregation and beyond. Nat. Rev. Cancer 14, 389–393 (2014). - PubMed
-
- Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, Ishikawa S, Sato-Otsubo A, Nagae G, Nishimoto A, Haferlach C, Nowak D, Sato Y, Alpermann T, Nagasaki M, Shimamura T, Tanaka H, Chiba K, Yamamoto R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Nakamaki T, Ishiyama K, Nolte F, Hofmann WK, Miyawaki S, Chiba S, Mori H, Nakauchi H, Koeffler HP, Aburatani H, Haferlach T, Shirahige K, Miyano S, Ogawa S, Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 45, 1232–1237 (2013). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
