

Heterozygous mutations in the C-terminal domain of COPA underlie a complex autoinflammatory syndrome
- PMID: 38175705
- PMCID: PMC10866661
- DOI: 10.1172/JCI163604
Heterozygous mutations in the C-terminal domain of COPA underlie a complex autoinflammatory syndrome
Abstract
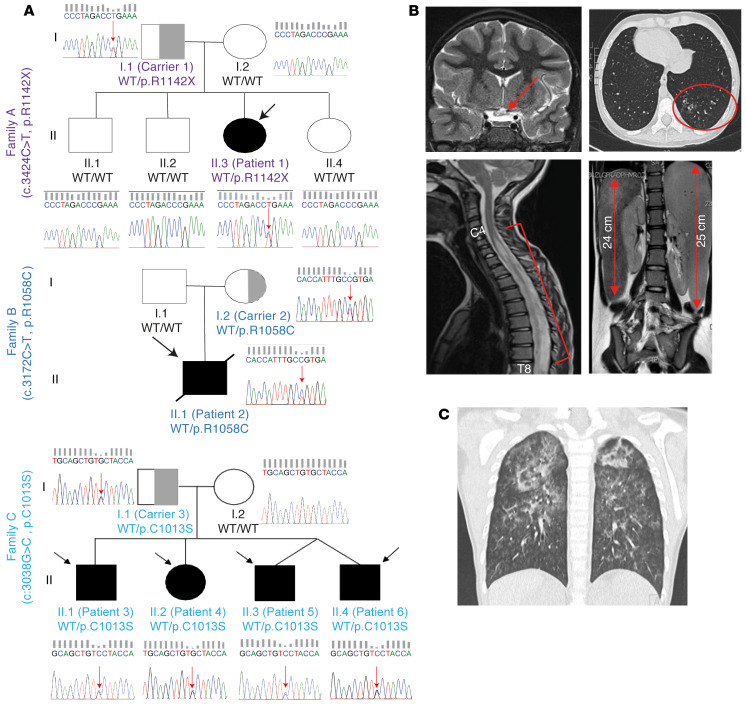
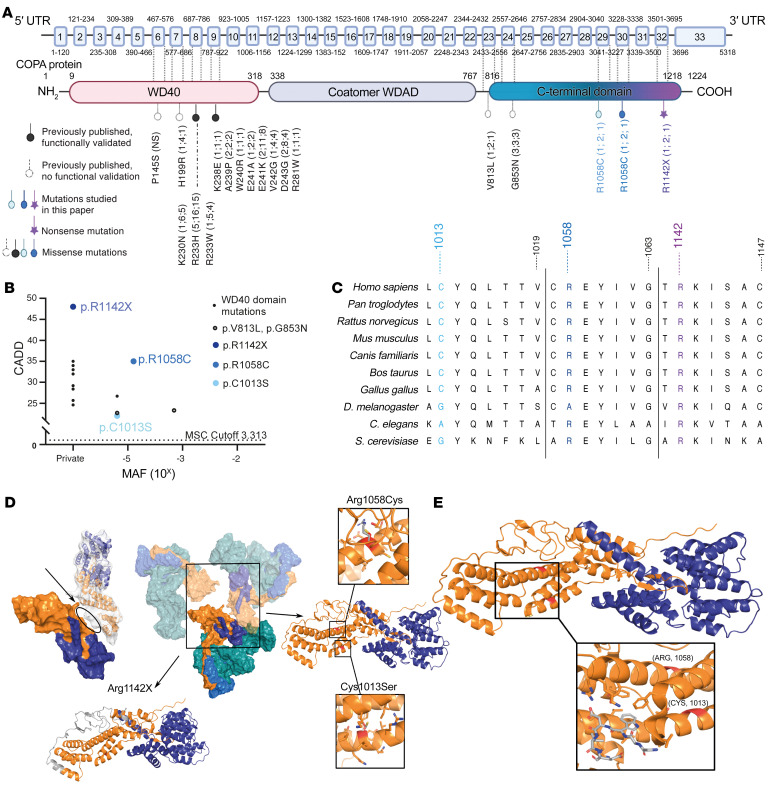
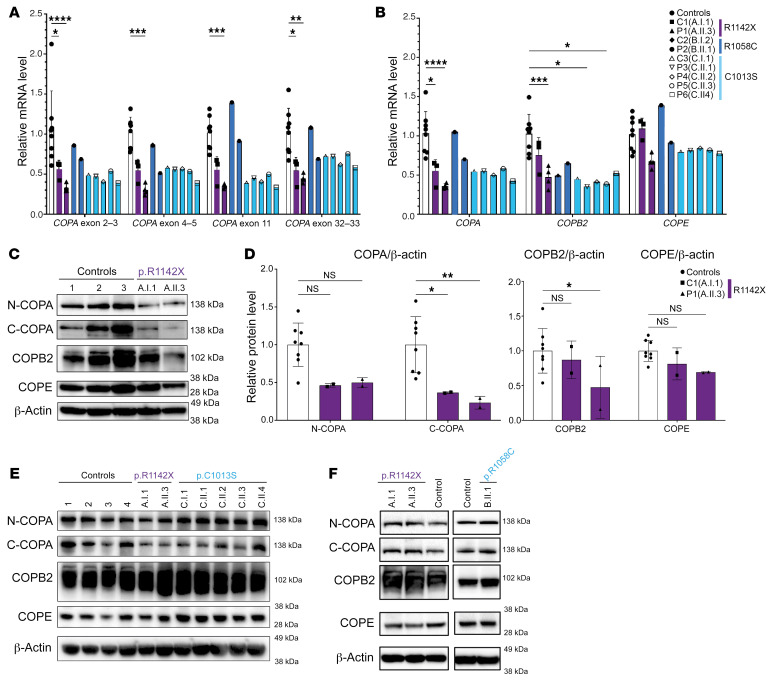
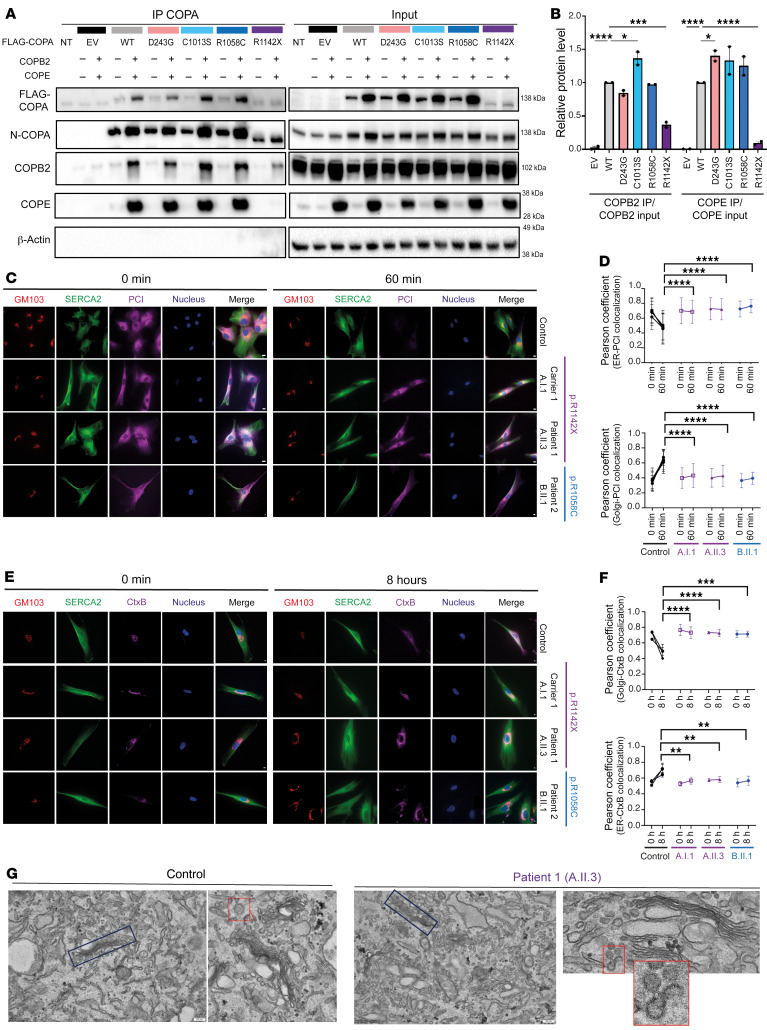
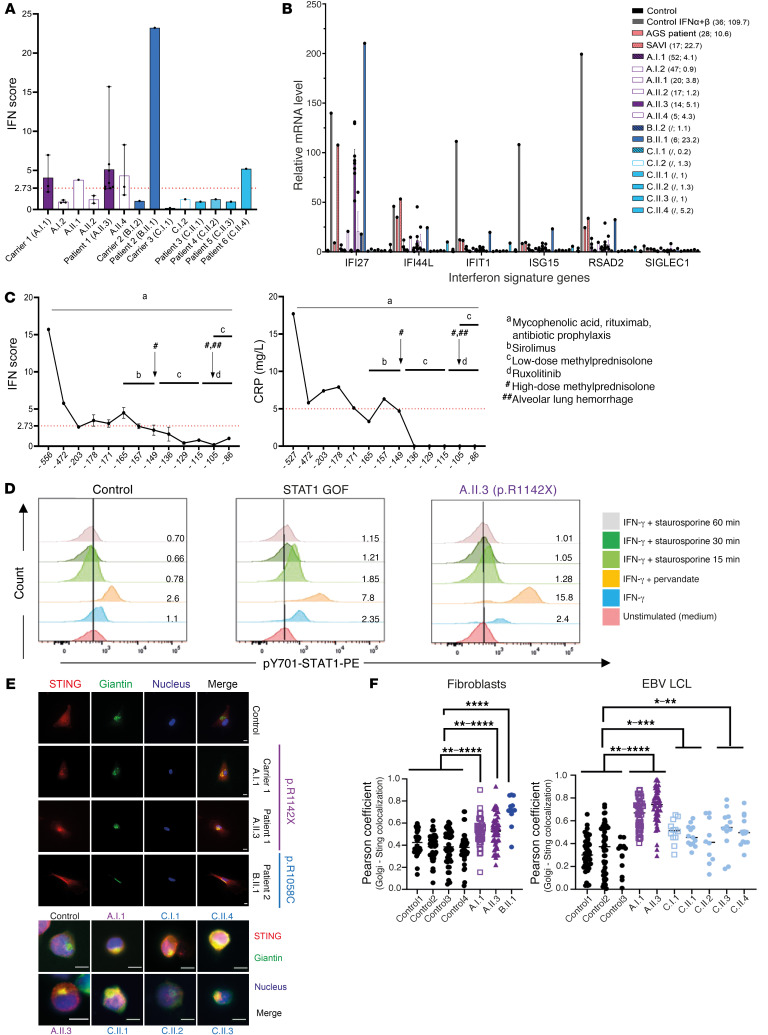
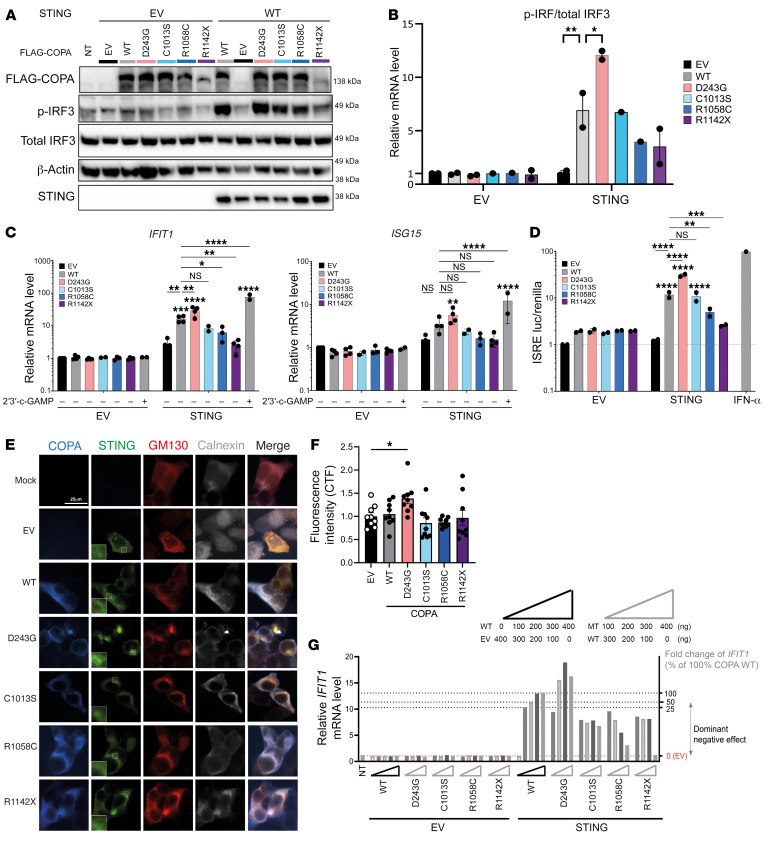
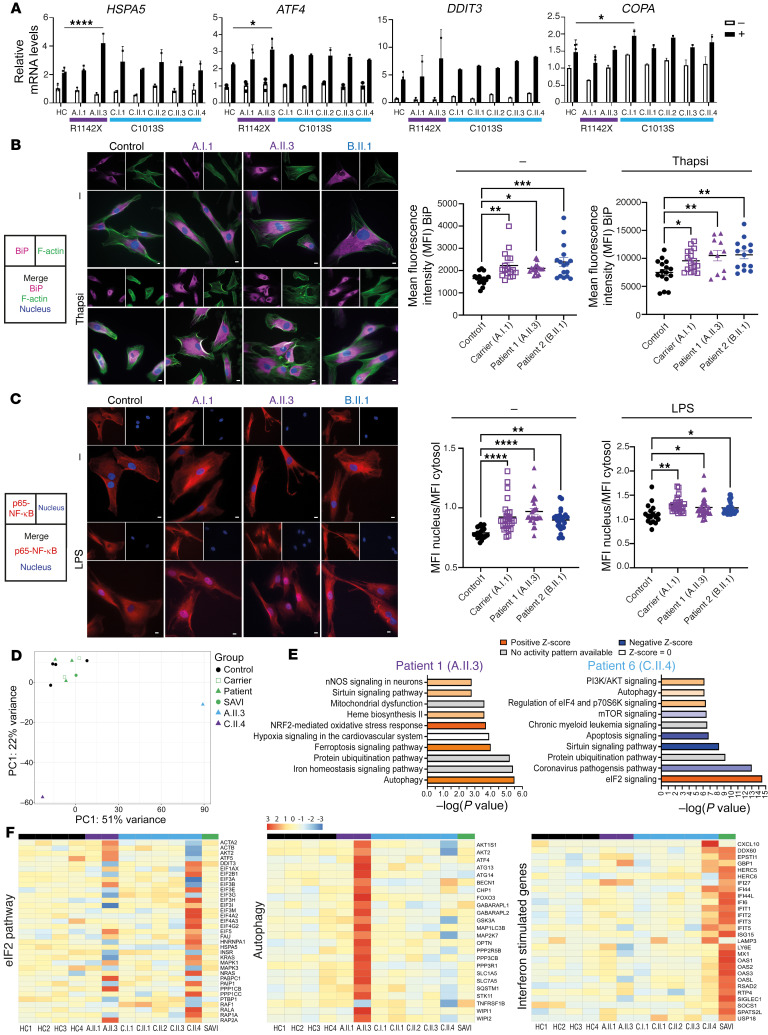
Mutations in the N-terminal WD40 domain of coatomer protein complex subunit α (COPA) cause a type I interferonopathy, typically characterized by alveolar hemorrhage, arthritis, and nephritis. We described 3 heterozygous mutations in the C-terminal domain (CTD) of COPA (p.C1013S, p.R1058C, and p.R1142X) in 6 children from 3 unrelated families with a similar syndrome of autoinflammation and autoimmunity. We showed that these CTD COPA mutations disrupt the integrity and the function of coat protein complex I (COPI). In COPAR1142X and COPAR1058C fibroblasts, we demonstrated that COPI dysfunction causes both an anterograde ER-to-Golgi and a retrograde Golgi-to-ER trafficking defect. The disturbed intracellular trafficking resulted in a cGAS/STING-dependent upregulation of the type I IFN signaling in patients and patient-derived cell lines, albeit through a distinct molecular mechanism in comparison with mutations in the WD40 domain of COPA. We showed that CTD COPA mutations induce an activation of ER stress and NF-κB signaling in patient-derived primary cell lines. These results demonstrate the importance of the integrity of the CTD of COPA for COPI function and homeostatic intracellular trafficking, essential to ER homeostasis. CTD COPA mutations result in disease by increased ER stress, disturbed intracellular transport, and increased proinflammatory signaling.
Keywords: Cell stress; Immunology; Innate immunity; Monogenic diseases.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
