

Dynamic metabolism of endothelial triglycerides protects against atherosclerosis in mice
- PMID: 38175710
- PMCID: PMC10866653
- DOI: 10.1172/JCI170453
Dynamic metabolism of endothelial triglycerides protects against atherosclerosis in mice
Abstract
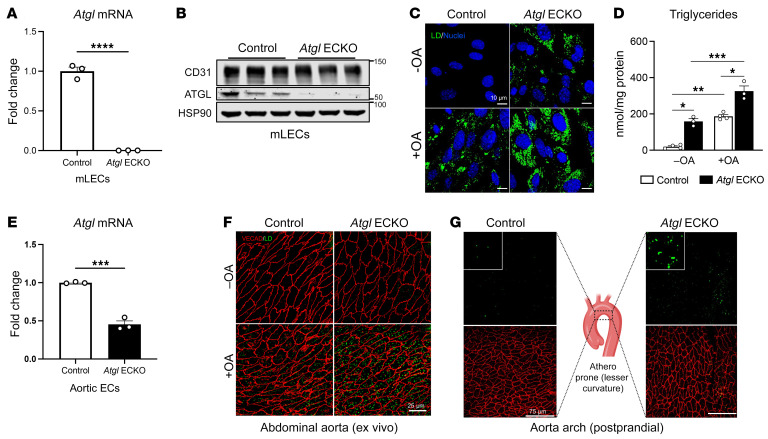
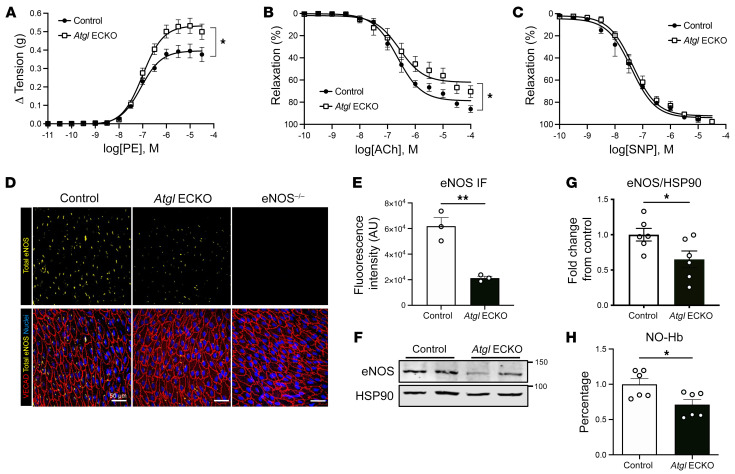
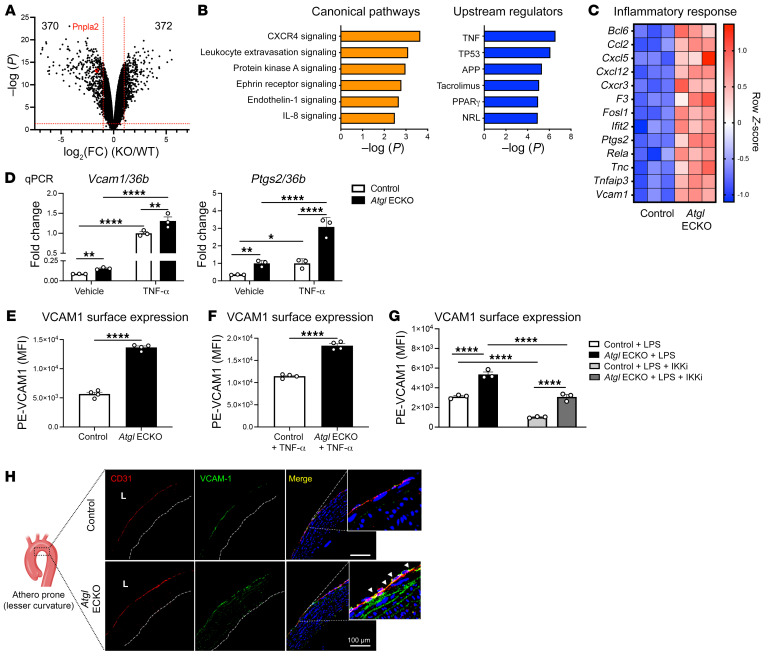
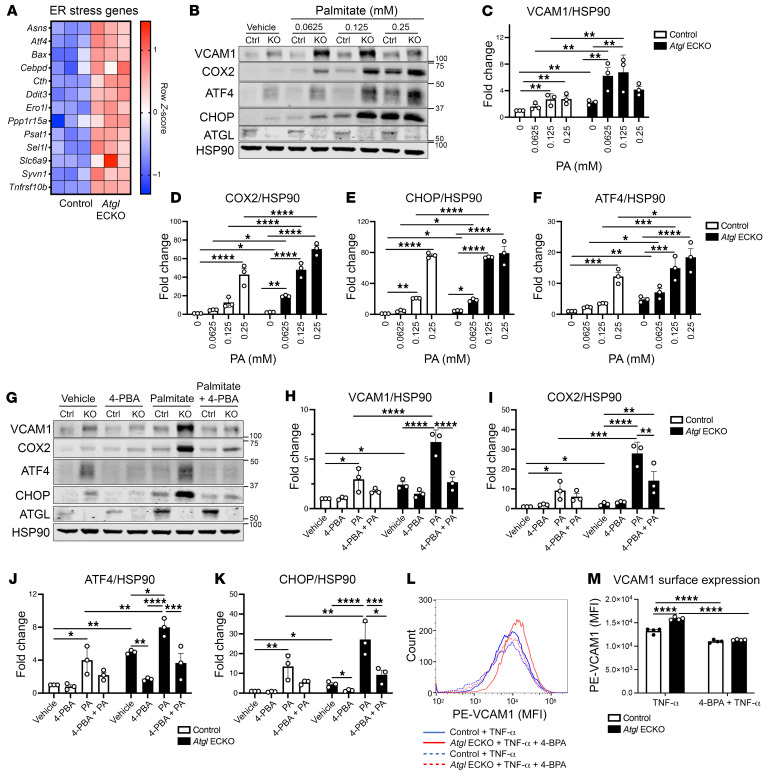
Blood vessels are continually exposed to circulating lipids, and elevation of ApoB-containing lipoproteins causes atherosclerosis. Lipoprotein metabolism is highly regulated by lipolysis, largely at the level of the capillary endothelium lining metabolically active tissues. How large blood vessels, the site of atherosclerotic vascular disease, regulate the flux of fatty acids (FAs) into triglyceride-rich (TG-rich) lipid droplets (LDs) is not known. In this study, we showed that deletion of the enzyme adipose TG lipase (ATGL) in the endothelium led to neutral lipid accumulation in vessels and impaired endothelial-dependent vascular tone and nitric oxide synthesis to promote endothelial dysfunction. Mechanistically, the loss of ATGL led to endoplasmic reticulum stress-induced inflammation in the endothelium. Consistent with this mechanism, deletion of endothelial ATGL markedly increased lesion size in a model of atherosclerosis. Together, these data demonstrate that the dynamics of FA flux through LD affects endothelial cell homeostasis and consequently large vessel function during normal physiology and in a chronic disease state.
Keywords: Atherosclerosis; Endothelial cells; Lipoproteins; Vascular Biology.
Figures
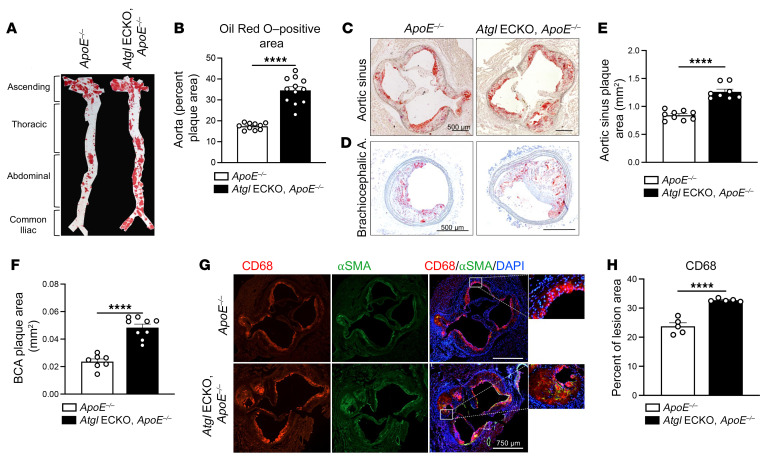
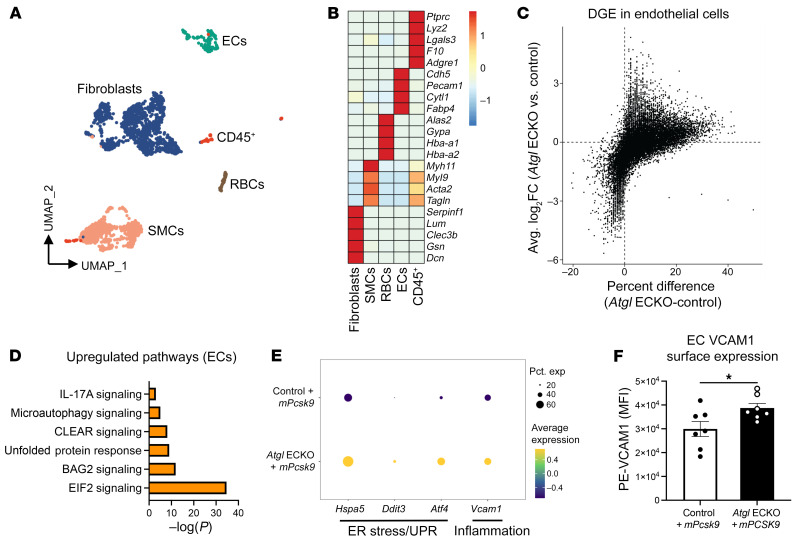
Comment in
- Lipid droplets in the endothelium: The missing link between metabolic syndrome and cardiovascular disease?
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
