

Network cartographs for interpretable visualizations
- PMID: 38177513
- PMCID: PMC10766564
- DOI: 10.1038/s43588-022-00199-z
Network cartographs for interpretable visualizations
Abstract
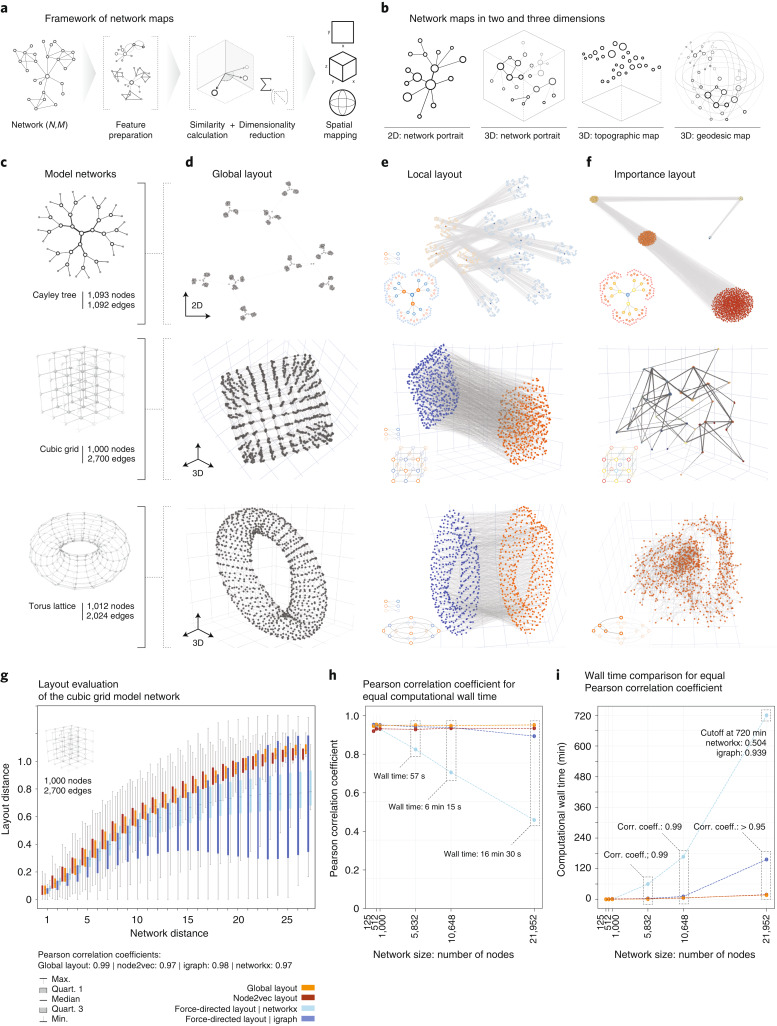
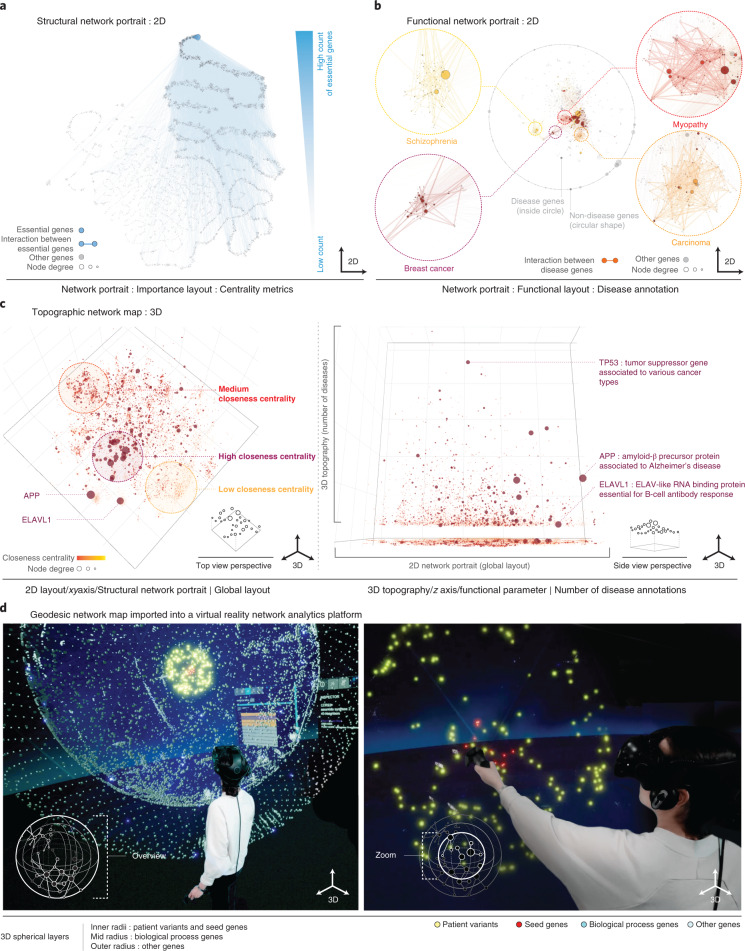
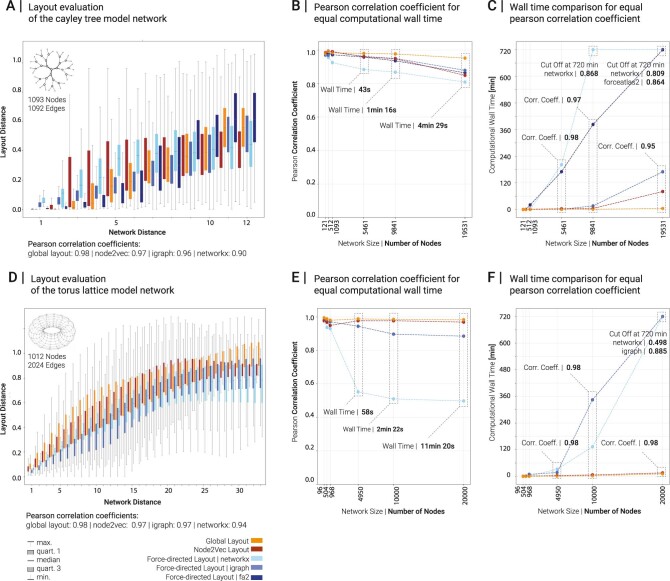
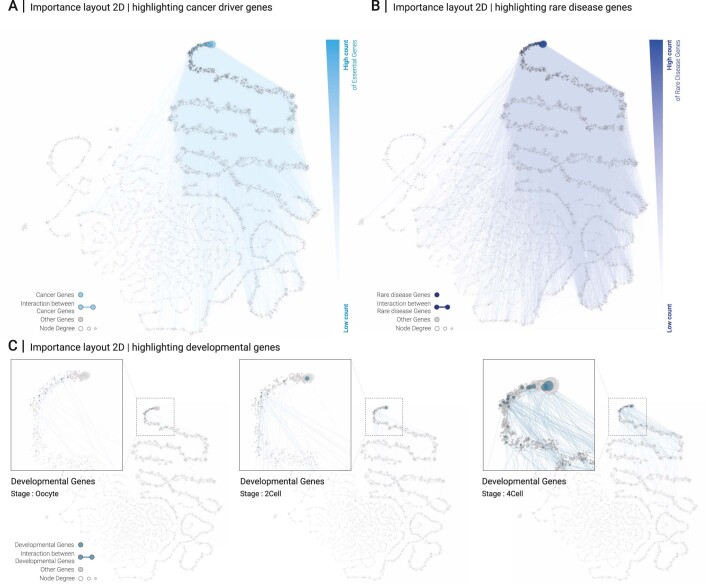
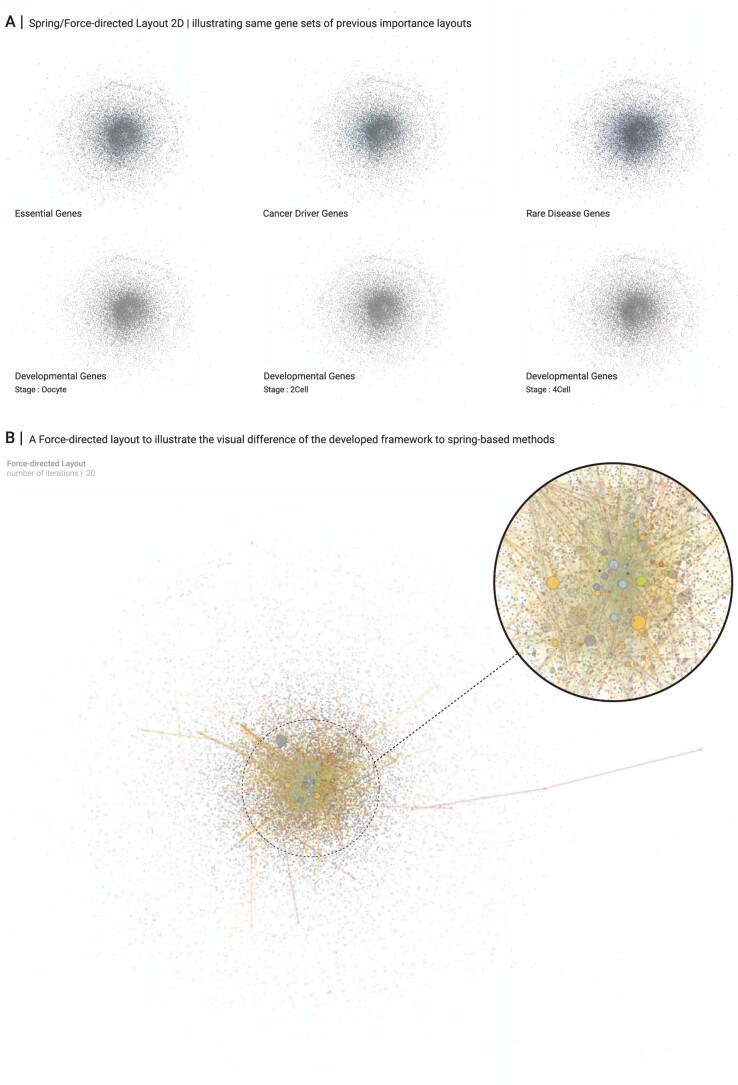
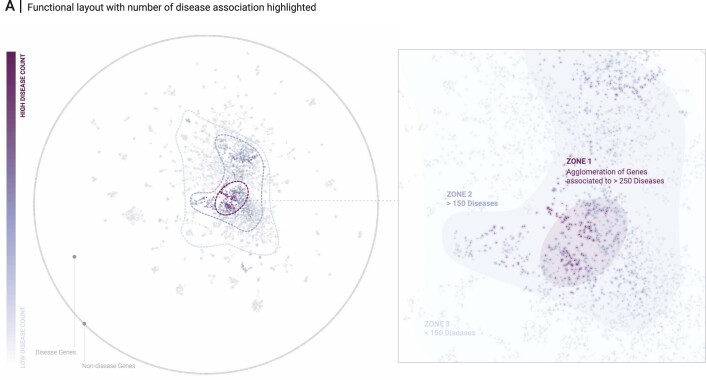
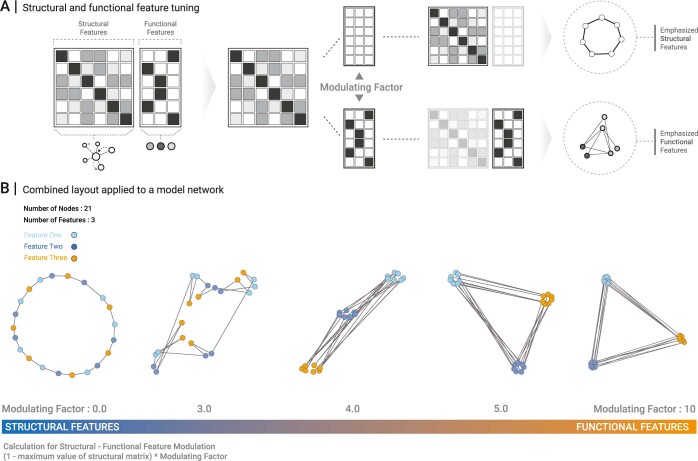
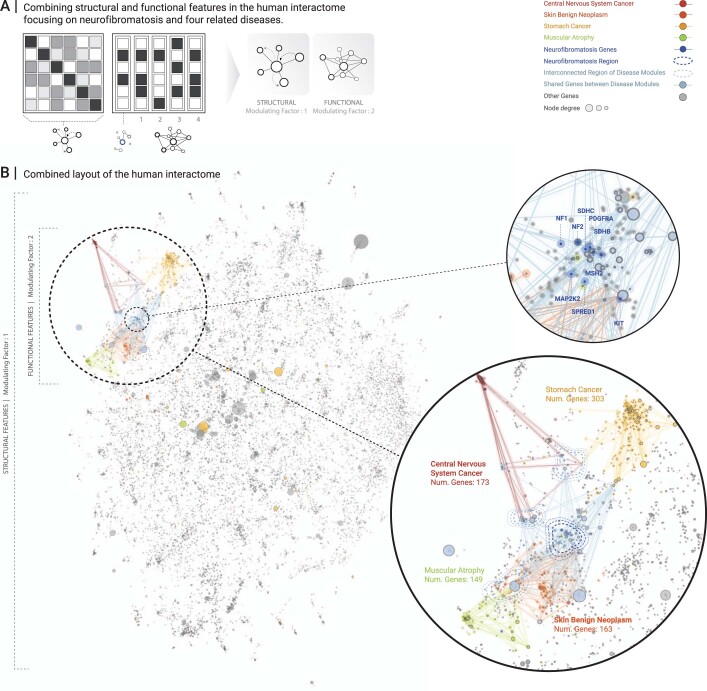
Networks offer an intuitive visual representation of complex systems. Important network characteristics can often be recognized by eye and, in turn, patterns that stand out visually often have a meaningful interpretation. In conventional network layout algorithms, however, the precise determinants of a node's position within a layout are difficult to decipher and to control. Here we propose an approach for directly encoding arbitrary structural or functional network characteristics into node positions. We introduce a series of two- and three-dimensional layouts, benchmark their efficiency for model networks, and demonstrate their power for elucidating structure-to-function relationships in large-scale biological networks.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Newman, M. Networks (Oxford Univ. Press, 2018).
Grants and funding
LinkOut - more resources
Full Text Sources
