

Using multi-scale genomics to associate poorly annotated genes with rare diseases
- PMID: 38178268
- PMCID: PMC10765705
- DOI: 10.1186/s13073-023-01276-2
Using multi-scale genomics to associate poorly annotated genes with rare diseases
Abstract
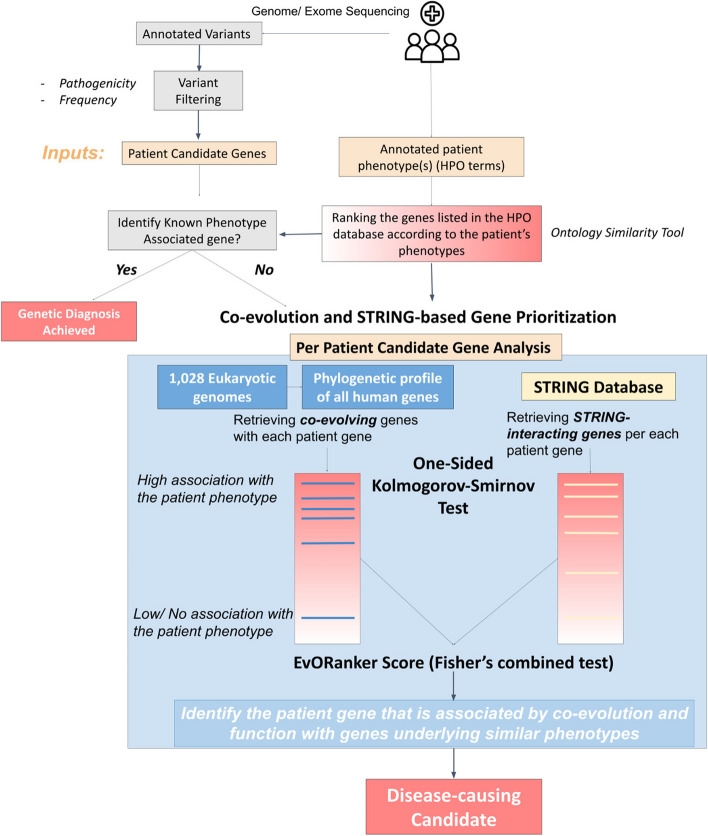
Background: Next-generation sequencing (NGS) has significantly transformed the landscape of identifying disease-causing genes associated with genetic disorders. However, a substantial portion of sequenced patients remains undiagnosed. This may be attributed not only to the challenges posed by harder-to-detect variants, such as non-coding and structural variations but also to the existence of variants in genes not previously associated with the patient's clinical phenotype. This study introduces EvORanker, an algorithm that integrates unbiased data from 1,028 eukaryotic genomes to link mutated genes to clinical phenotypes.
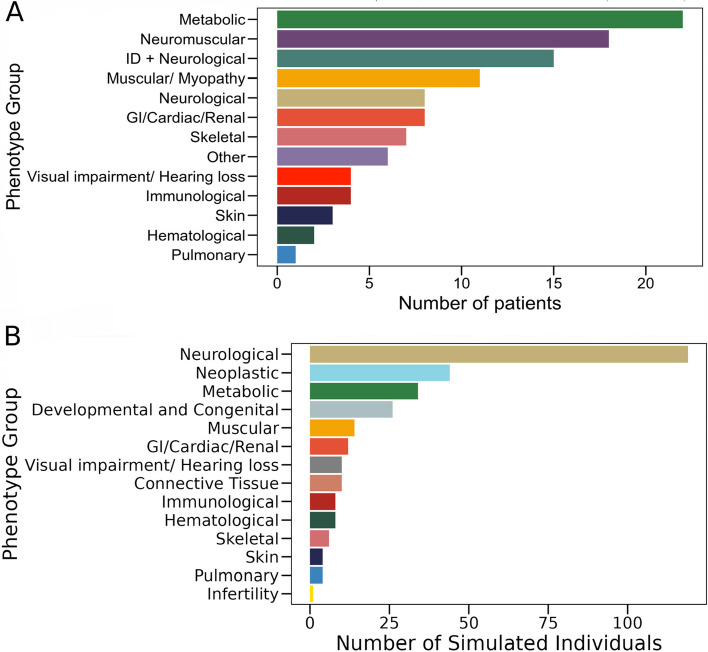
Methods: EvORanker utilizes clinical data, multi-scale phylogenetic profiling, and other omics data to prioritize disease-associated genes. It was evaluated on solved exomes and simulated genomes, compared with existing methods, and applied to 6260 knockout genes with mouse phenotypes lacking human associations. Additionally, EvORanker was made accessible as a user-friendly web tool.
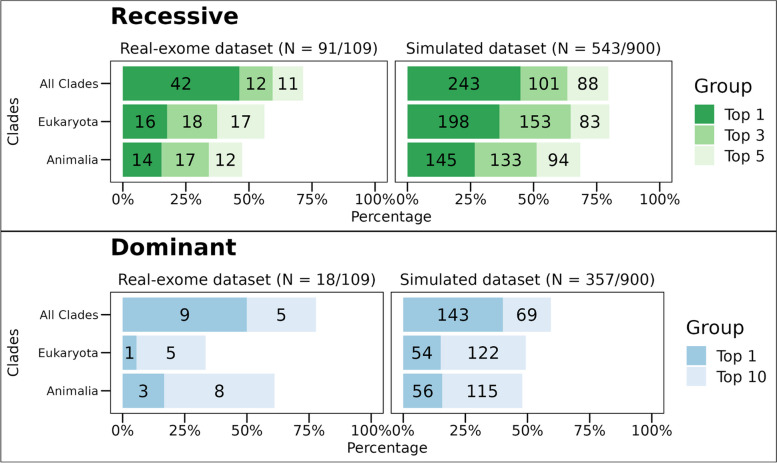
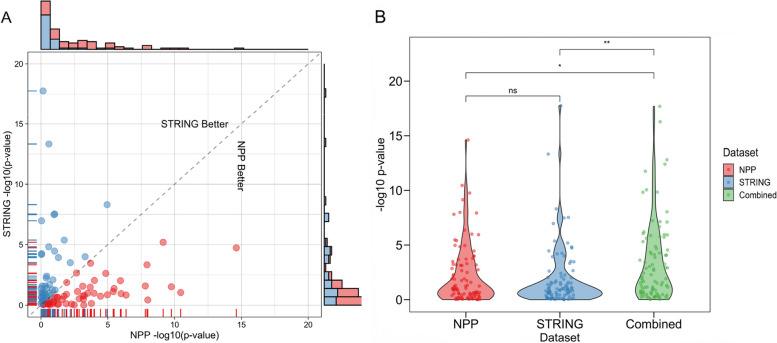
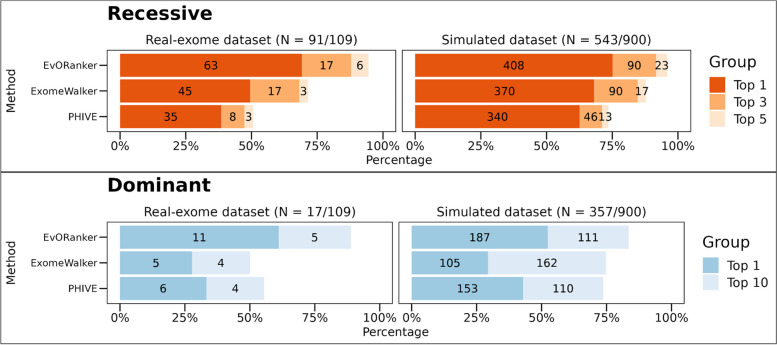
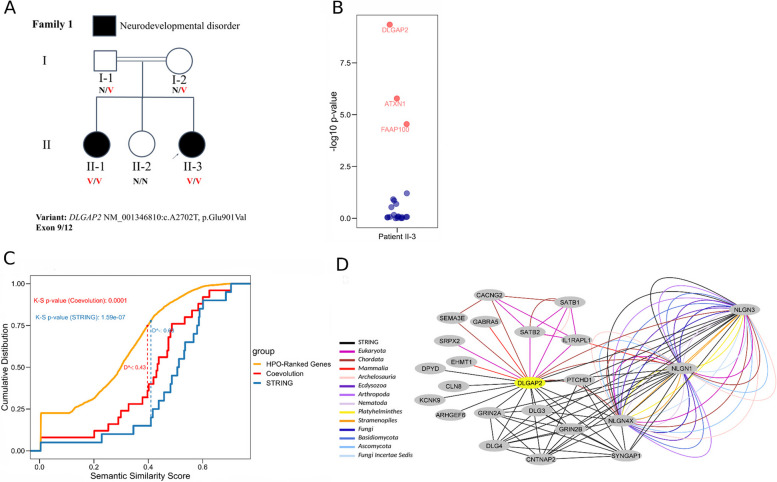
Results: In the analyzed exomic cohort, EvORanker accurately identified the "true" disease gene as the top candidate in 69% of cases and within the top 5 candidates in 95% of cases, consistent with results from the simulated dataset. Notably, EvORanker outperformed existing methods, particularly for poorly annotated genes. In the case of the 6260 knockout genes with mouse phenotypes, EvORanker linked 41% of these genes to observed human disease phenotypes. Furthermore, in two unsolved cases, EvORanker successfully identified DLGAP2 and LPCAT3 as disease candidates for previously uncharacterized genetic syndromes.
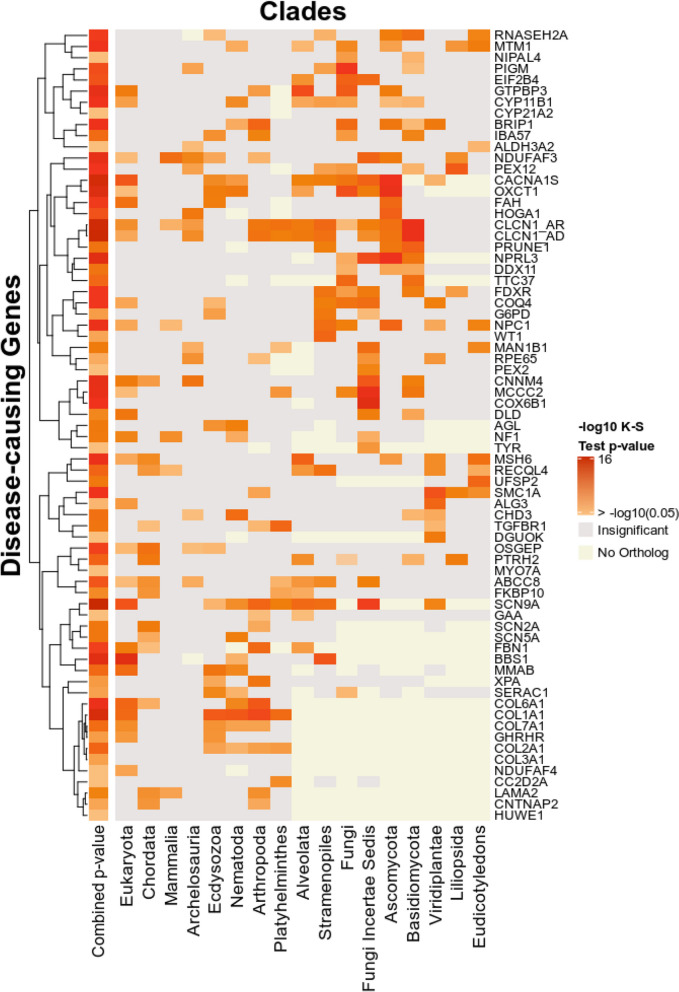
Conclusions: We highlight clade-based phylogenetic profiling as a powerful systematic approach for prioritizing potential disease genes. Our study showcases the efficacy of EvORanker in associating poorly annotated genes to disease phenotypes observed in patients. The EvORanker server is freely available at https://ccanavati.shinyapps.io/EvORanker/ .
Keywords: DLGAP2; EvORanker; Gene-based prioritization; LPCAT3.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD). 2023. https://omim.org/. Accessed 18 Sept 2023.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
