

Structural coverage of the human interactome
- PMID: 38180828
- PMCID: PMC10768791
- DOI: 10.1093/bib/bbad496
Structural coverage of the human interactome
Abstract
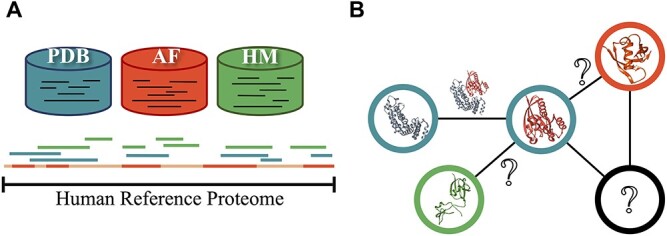
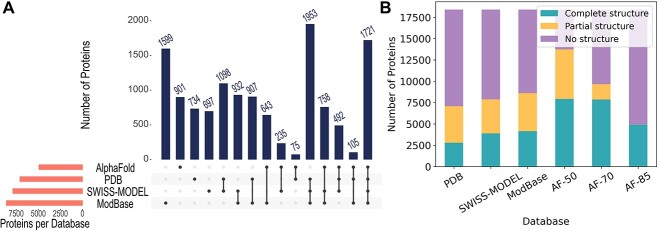
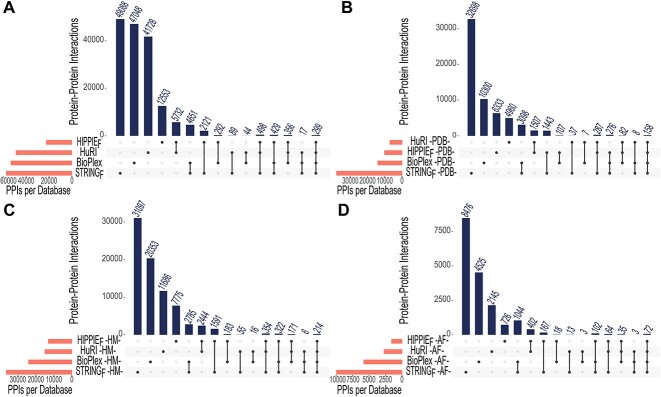
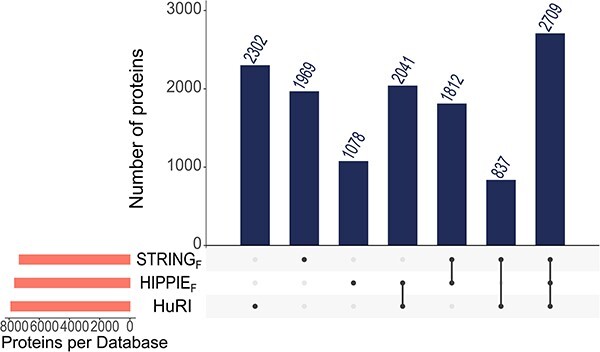
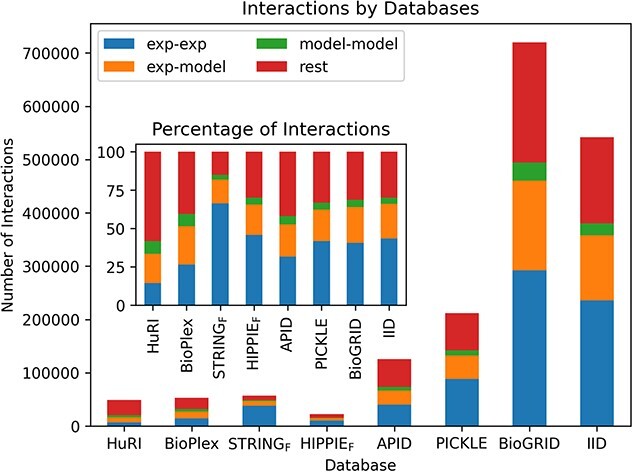
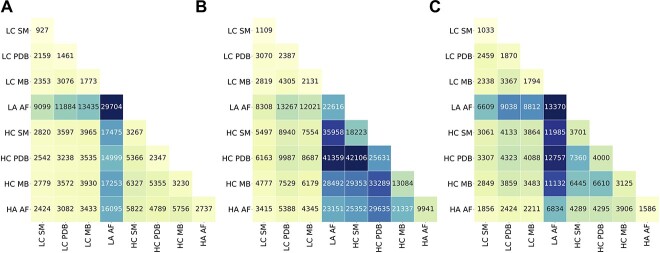
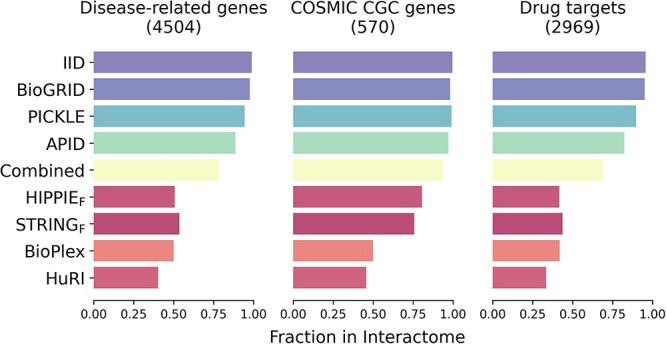
Complex biological processes in cells are embedded in the interactome, representing the complete set of protein-protein interactions. Mapping and analyzing the protein structures are essential to fully comprehending these processes' molecular details. Therefore, knowing the structural coverage of the interactome is important to show the current limitations. Structural modeling of protein-protein interactions requires accurate protein structures. In this study, we mapped all experimental structures to the reference human proteome. Later, we found the enrichment in structural coverage when complementary methods such as homology modeling and deep learning (AlphaFold) were included. We then collected the interactions from the literature and databases to form the reference human interactome, resulting in 117 897 non-redundant interactions. When we analyzed the structural coverage of the interactome, we found that the number of experimentally determined protein complex structures is scarce, corresponding to 3.95% of all binary interactions. We also analyzed known and modeled structures to potentially construct the structural interactome with a docking method. Our analysis showed that 12.97% of the interactions from HuRI and 73.62% and 32.94% from the filtered versions of STRING and HIPPIE could potentially be modeled with high structural coverage or accuracy, respectively. Overall, this paper provides an overview of the current state of structural coverage of the human proteome and interactome.
Keywords: AlphaFold2; PDB; homology modeling databases; human interactome; human proteome; protein complexes; structural coverage.
© The Author(s) 2024. Published by Oxford University Press.
Figures
Similar articles
-
CM2D3: Furnishing the Human Interactome with Structural Models of Protein Complexes Derived by Comparative Modeling and Docking.J Mol Biol. 2023 Jul 15;435(14):168055. doi: 10.1016/j.jmb.2023.168055. Epub 2023 Mar 21. J Mol Biol. 2023. PMID: 36958605
-
How AlphaFold2 shaped the structural coverage of the human transmembrane proteome.Sci Rep. 2023 Nov 20;13(1):20283. doi: 10.1038/s41598-023-47204-7. Sci Rep. 2023. PMID: 37985809 Free PMC article.
-
The social and structural architecture of the yeast protein interactome.Nature. 2023 Dec;624(7990):192-200. doi: 10.1038/s41586-023-06739-5. Epub 2023 Nov 15. Nature. 2023. PMID: 37968396 Free PMC article.
-
Low-resolution structural modeling of protein interactome.Curr Opin Struct Biol. 2013 Apr;23(2):198-205. doi: 10.1016/j.sbi.2012.12.003. Epub 2013 Jan 5. Curr Opin Struct Biol. 2013. PMID: 23294579 Free PMC article. Review.
-
Protein-protein docking: from interaction to interactome.Biophys J. 2014 Oct 21;107(8):1785-1793. doi: 10.1016/j.bpj.2014.08.033. Biophys J. 2014. PMID: 25418159 Free PMC article. Review.
Cited by
-
NetREm: Network Regression Embeddings reveal cell-type transcription factor coordination for gene regulation.Bioinform Adv. 2024 Dec 20;5(1):vbae206. doi: 10.1093/bioadv/vbae206. eCollection 2025. Bioinform Adv. 2024. PMID: 40260118 Free PMC article.
-
Analysis of Modular Hub Genes and Therapeutic Targets across Stages of Non-Small Cell Lung Cancer Transcriptome.Genes (Basel). 2024 Sep 25;15(10):1248. doi: 10.3390/genes15101248. Genes (Basel). 2024. PMID: 39457373 Free PMC article.
-
Special Issue "Deployment of Proteomics Approaches in Biomedical Research".Int J Mol Sci. 2024 Jan 31;25(3):1717. doi: 10.3390/ijms25031717. Int J Mol Sci. 2024. PMID: 38338994 Free PMC article.
-
Discovery of a Novel Chemo-Type for TAAR1 Agonism via Molecular Modeling.Molecules. 2024 Apr 11;29(8):1739. doi: 10.3390/molecules29081739. Molecules. 2024. PMID: 38675561 Free PMC article.
References
-
- Garland W, Benezra R, Chaudhary J. Chapter fifteen—targeting protein–protein interactions to treat cancer—recent progress and future directions. In: Desai MC (ed). Annual Reports in Medicinal Chemistry, Vol. 48., Massachusetts: Academic Press, 2013, 227–45.
-
- Mosca R, Céol A, Aloy P. Interactome3D: adding structural details to protein networks. Nat Methods 2013;10(1):47–53. - PubMed
