

Clinical, laboratory, and molecular characteristics of a cohort of children with hemoglobinopathy S/beta-thalassemia
- PMID: 38182466
- PMCID: PMC11150486
- DOI: 10.1016/j.htct.2023.11.002
Clinical, laboratory, and molecular characteristics of a cohort of children with hemoglobinopathy S/beta-thalassemia
Abstract
Introduction: Hemoglobinopathy Sβ-thalassemia (HbSβ-thal) has a wide range of clinical and laboratory severity. There is limited information on the natural history of HbSβ-thal and its modulating factors. We described the molecular, hematological, and clinical characteristics of a cohort of children with HbSβ-thal and estimated its incidence in Minas Gerais, Brazil.
Methods: Laboratory and clinical data were retrieved from medical records. Molecular analysis was performed by HBB gene sequencing, PCR-RFLP, gap-PCR, and MLPA.
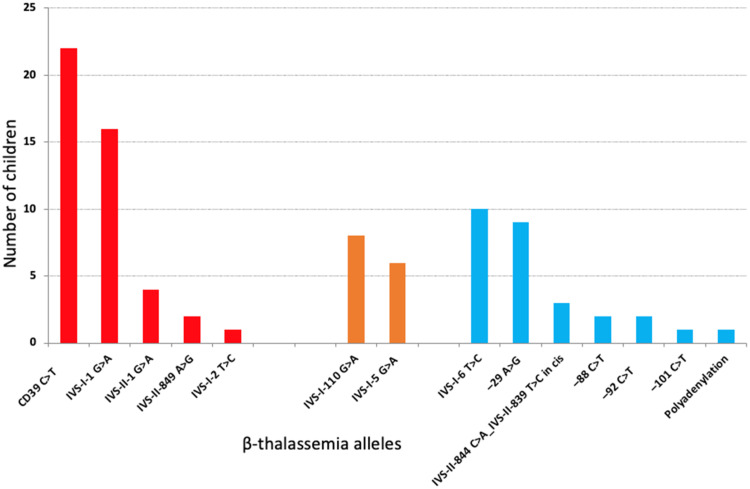
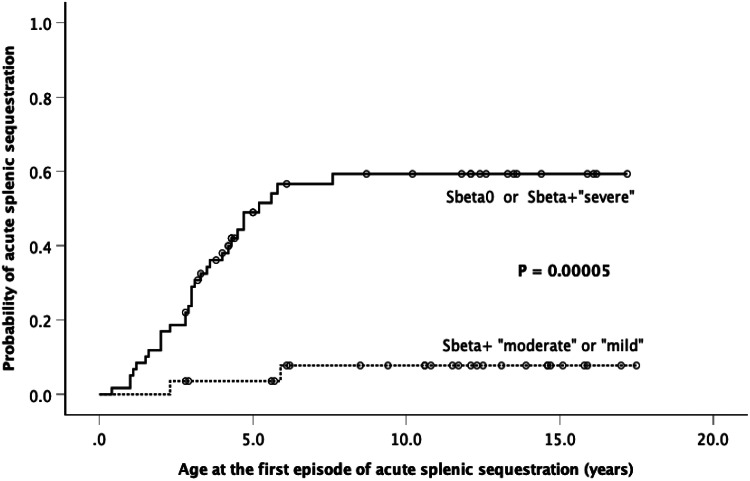
Results: Eighty-nine children were included in the study. Fourteen alleles of β-thal mutations were identified. The incidence of HbSβ-thal in the state was 1 per 22,250 newborns. The most common βS-haplotypes were CAR and Benin. The most frequent βthal-haplotypes were V, II, and I. Coexistence of 3.7 kb HBA1/HBA2 deletion was present in 21.3 % of children. β-thalassemia mutations were associated with several clinical and laboratory features. In general, the incidence of clinical events per 100 patient-years was similar for children with HbSβ0-thal, IVS-I-5 G>A, and IVS-I-110 G>A. Children with HbSβ+-intermediate phenotypes had a more severe laboratory and clinical profile when compared with those with HbSβ+-mild ones. βS-haplotypes and α-thalassemia did not meaningfully influence the phenotype of children with HbSβ-thal.
Conclusion: The early identification of β-thalassemia alleles may help the clinical management of these children.
Keywords: Alpha-thalassemia; Hemoglobin Sβ(+)-thalassemia; Hemoglobin Sβ(0)-thalassemia; Hemoglobin Sβ-thalassemia; Sickle cell disease.
Copyright © 2023 Associação Brasileira de Hematologia, Hemoterapia e Terapia Celular. Published by Elsevier España, S.L.U. All rights reserved.
Conflict of interest statement
Conflicts of interest All authors declare that they have no conflict of interest.
Figures
Similar articles
-
Ten Years of Routine α- and β-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations.Hemoglobin. 2016;40(2):75-84. doi: 10.3109/03630269.2015.1113990. Epub 2015 Dec 4. Hemoglobin. 2016. PMID: 26635043 Review.
-
Hb S/β-Thalassemia in the REDS-III Brazil Sickle Cell Disease Cohort: Clinical, Laboratory and Molecular Characteristics.Hemoglobin. 2020 Jan;44(1):1-9. doi: 10.1080/03630269.2020.1731530. Epub 2020 Mar 16. Hemoglobin. 2020. PMID: 32172616 Free PMC article.
-
Molecular analysis of complex cases of alpha- and beta-thalassemia in Mexican mestizo patients with microcytosis and hypochromia reveals two novel alpha(0) -thalassemia deletions - -(Mex1) and - -(Mex2).Int J Lab Hematol. 2016 Oct;38(5):535-42. doi: 10.1111/ijlh.12536. Epub 2016 Jun 24. Int J Lab Hematol. 2016. PMID: 27339814
-
Molecular and Hematological Analysis of Alpha- and Beta-Thalassemia in a Cohort of Mexican Patients.Genet Test Mol Biomarkers. 2021 Mar;25(3):247-252. doi: 10.1089/gtmb.2020.0276. Genet Test Mol Biomarkers. 2021. PMID: 33734896
-
β-Thalassemia Haplotypes in Romania in the Context of Genetic Mixing in the Mediterranean Area.Hemoglobin. 2016;40(2):85-96. doi: 10.3109/03630269.2015.1124113. Epub 2015 Dec 29. Hemoglobin. 2016. PMID: 26711012 Review.
Cited by
-
Are we securing our future workforce of physician-scientists in hematology?Hematol Transfus Cell Ther. 2024 Apr-Jun;46(2):111-112. doi: 10.1016/j.htct.2024.05.001. Hematol Transfus Cell Ther. 2024. PMID: 38789158 Free PMC article. No abstract available.
-
Advanced molecular approaches to thalassemia disorder and the selection of molecular-level diagnostic testing in resource-limited settings.Hematol Transfus Cell Ther. 2025 Jul-Sep;47(3):103860. doi: 10.1016/j.htct.2025.103860. Epub 2025 Jun 14. Hematol Transfus Cell Ther. 2025. PMID: 40523316 Free PMC article. Review.
References
-
- Cançado R., Jesus J.A. Sickle cell disease in Brazil. Rev Bras Hematol Hemoter. 2007;29(3):204–6. 10.1590/S1516-84842007000300002. - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous
