

STEM enables mapping of single-cell and spatial transcriptomics data with transfer learning
- PMID: 38184694
- PMCID: PMC10771471
- DOI: 10.1038/s42003-023-05640-1
STEM enables mapping of single-cell and spatial transcriptomics data with transfer learning
Abstract
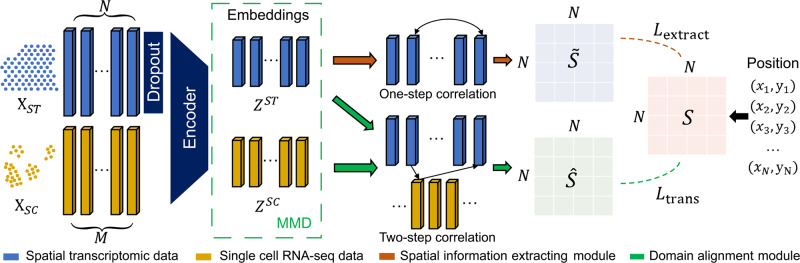
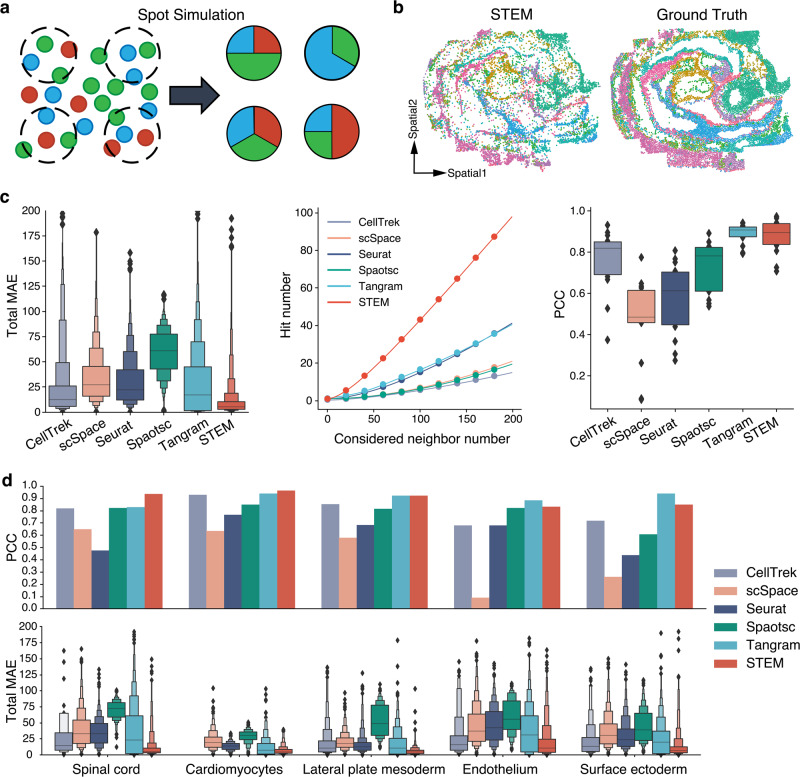
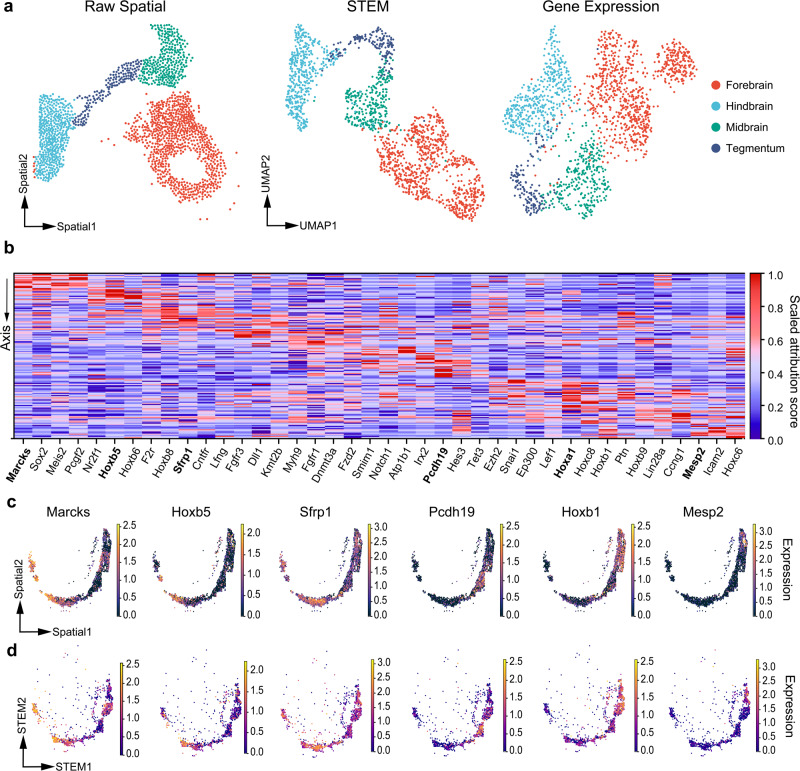
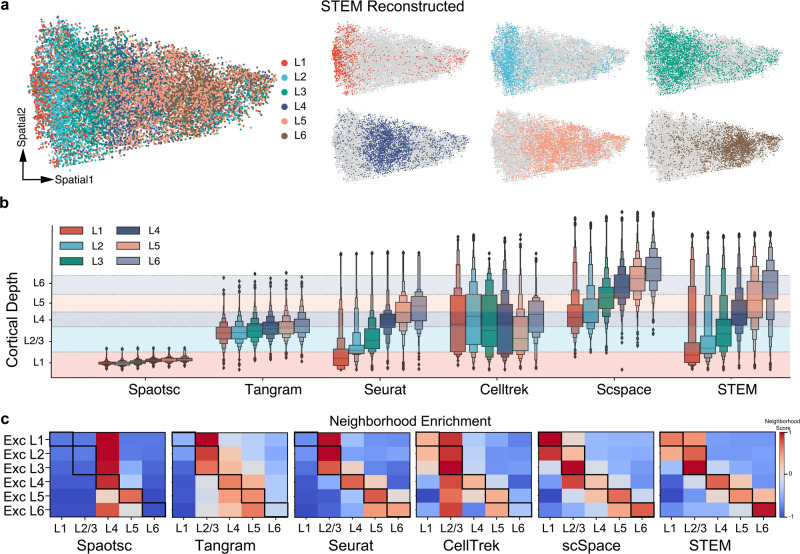
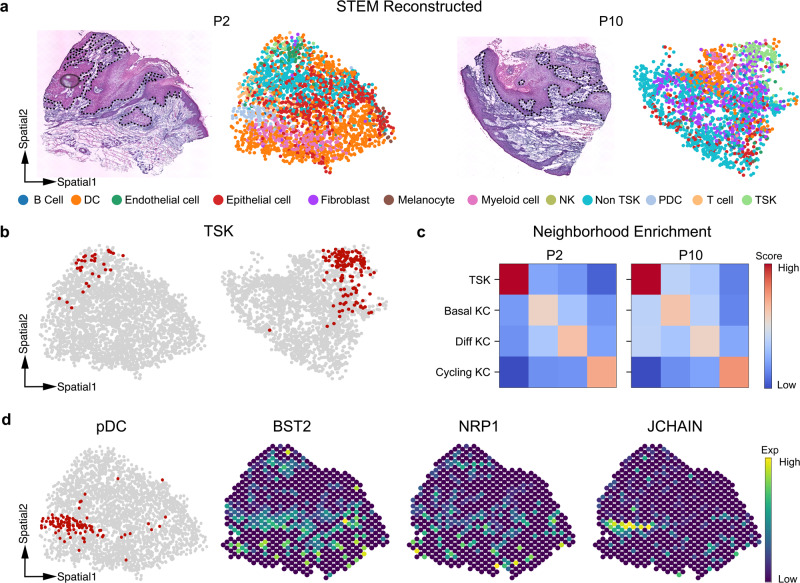
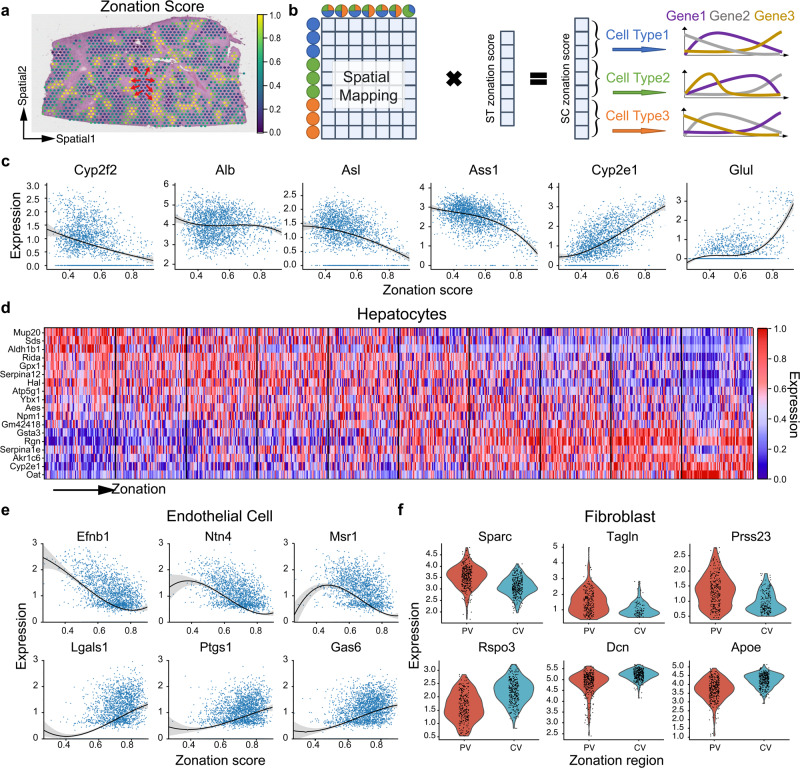
Profiling spatial variations of cellular composition and transcriptomic characteristics is important for understanding the physiology and pathology of tissues. Spatial transcriptomics (ST) data depict spatial gene expression but the currently dominating high-throughput technology is yet not at single-cell resolution. Single-cell RNA-sequencing (SC) data provide high-throughput transcriptomic information at the single-cell level but lack spatial information. Integrating these two types of data would be ideal for revealing transcriptomic landscapes at single-cell resolution. We develop the method STEM (SpaTially aware EMbedding) for this purpose. It uses deep transfer learning to encode both ST and SC data into a unified spatially aware embedding space, and then uses the embeddings to infer SC-ST mapping and predict pseudo-spatial adjacency between cells in SC data. Semi-simulation and real data experiments verify that the embeddings preserved spatial information and eliminated technical biases between SC and ST data. We apply STEM to human squamous cell carcinoma and hepatic lobule datasets to uncover the localization of rare cell types and reveal cell-type-specific gene expression variation along a spatial axis. STEM is powerful for mapping SC and ST data to build single-cell level spatial transcriptomic landscapes, and can provide mechanistic insights into the spatial heterogeneity and microenvironments of tissues.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Computational solutions for spatial transcriptomics.Comput Struct Biotechnol J. 2022 Sep 1;20:4870-4884. doi: 10.1016/j.csbj.2022.08.043. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36147664 Free PMC article. Review.
-
SpatialcoGCN: deconvolution and spatial information-aware simulation of spatial transcriptomics data via deep graph co-embedding.Brief Bioinform. 2024 Mar 27;25(3):bbae130. doi: 10.1093/bib/bbae130. Brief Bioinform. 2024. PMID: 38557675 Free PMC article.
-
Spatial-ID: a cell typing method for spatially resolved transcriptomics via transfer learning and spatial embedding.Nat Commun. 2022 Dec 10;13(1):7640. doi: 10.1038/s41467-022-35288-0. Nat Commun. 2022. PMID: 36496406 Free PMC article.
-
SD2: spatially resolved transcriptomics deconvolution through integration of dropout and spatial information.Bioinformatics. 2022 Oct 31;38(21):4878-4884. doi: 10.1093/bioinformatics/btac605. Bioinformatics. 2022. PMID: 36063455 Free PMC article.
-
A comprehensive comparison on cell-type composition inference for spatial transcriptomics data.Brief Bioinform. 2022 Jul 18;23(4):bbac245. doi: 10.1093/bib/bbac245. Brief Bioinform. 2022. PMID: 35753702 Free PMC article. Review.
Cited by
-
Transfer learning of multicellular organization via single-cell and spatial transcriptomics.PLoS Comput Biol. 2025 Apr 21;21(4):e1012991. doi: 10.1371/journal.pcbi.1012991. eCollection 2025 Apr. PLoS Comput Biol. 2025. PMID: 40258090 Free PMC article.
-
SELF-Former: multi-scale gene filtration transformer for single-cell spatial reconstruction.Brief Bioinform. 2024 Sep 23;25(6):bbae523. doi: 10.1093/bib/bbae523. Brief Bioinform. 2024. PMID: 39413798 Free PMC article.
-
Refinement strategies for Tangram for reliable single-cell to spatial mapping.Bioinformatics. 2025 Jul 1;41(Supplement_1):i552-i560. doi: 10.1093/bioinformatics/btaf194. Bioinformatics. 2025. PMID: 40662790 Free PMC article.
-
Deep learning in integrating spatial transcriptomics with other modalities.Brief Bioinform. 2024 Nov 22;26(1):bbae719. doi: 10.1093/bib/bbae719. Brief Bioinform. 2024. PMID: 39800876 Free PMC article. Review.
-
Building a learnable universal coordinate system for single-cell atlas with a joint-VAE model.Commun Biol. 2024 Aug 12;7(1):977. doi: 10.1038/s42003-024-06564-0. Commun Biol. 2024. PMID: 39134617 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
