

Rare copy-number variants as modulators of common disease susceptibility
- PMID: 38185688
- PMCID: PMC10773105
- DOI: 10.1186/s13073-023-01265-5
Rare copy-number variants as modulators of common disease susceptibility
Abstract
Background: Copy-number variations (CNVs) have been associated with rare and debilitating genomic disorders (GDs) but their impact on health later in life in the general population remains poorly described.
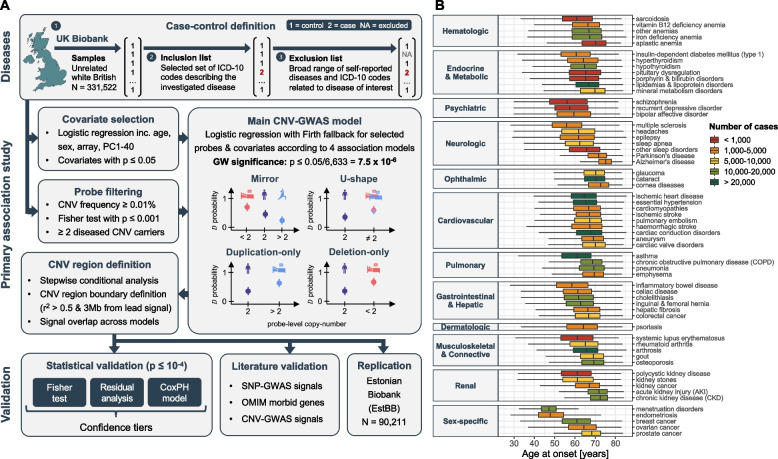
Methods: Assessing four modes of CNV action, we performed genome-wide association scans (GWASs) between the copy-number of CNV-proxy probes and 60 curated ICD-10 based clinical diagnoses in 331,522 unrelated white British UK Biobank (UKBB) participants with replication in the Estonian Biobank.
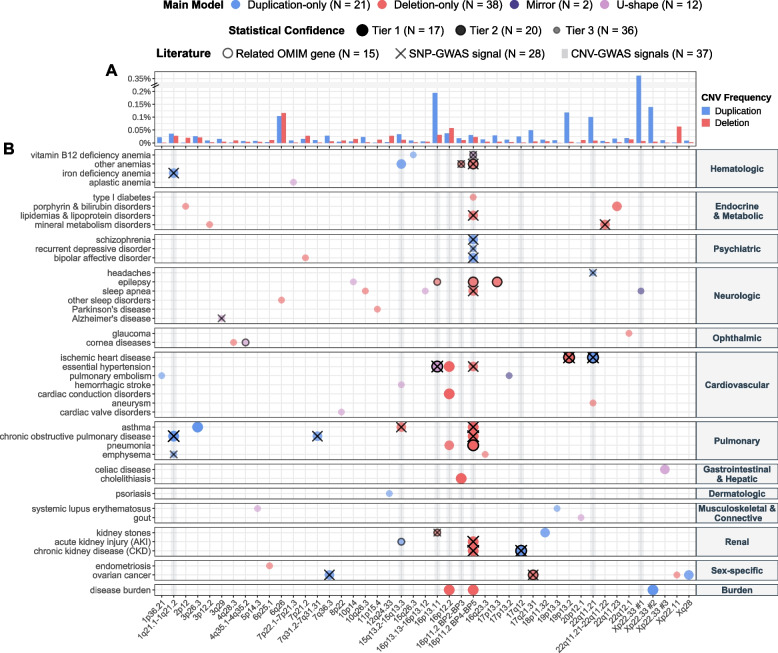
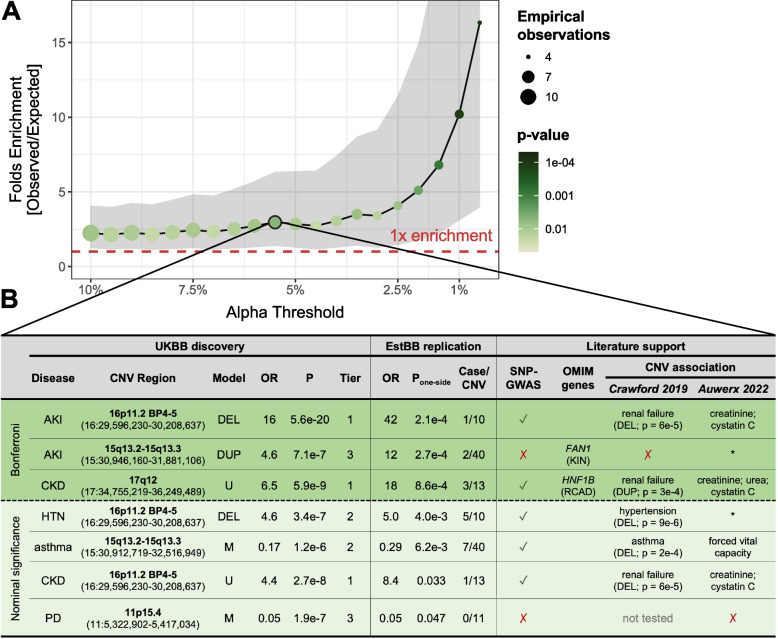
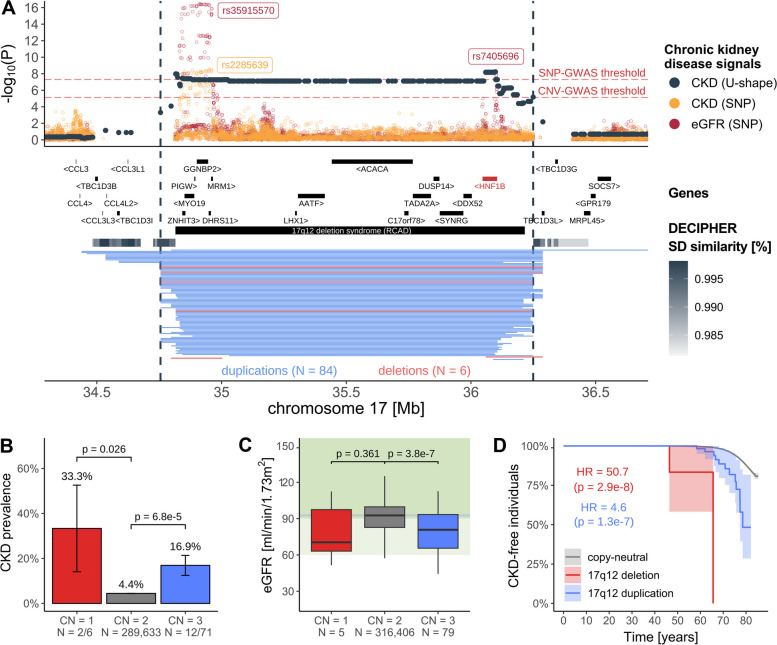
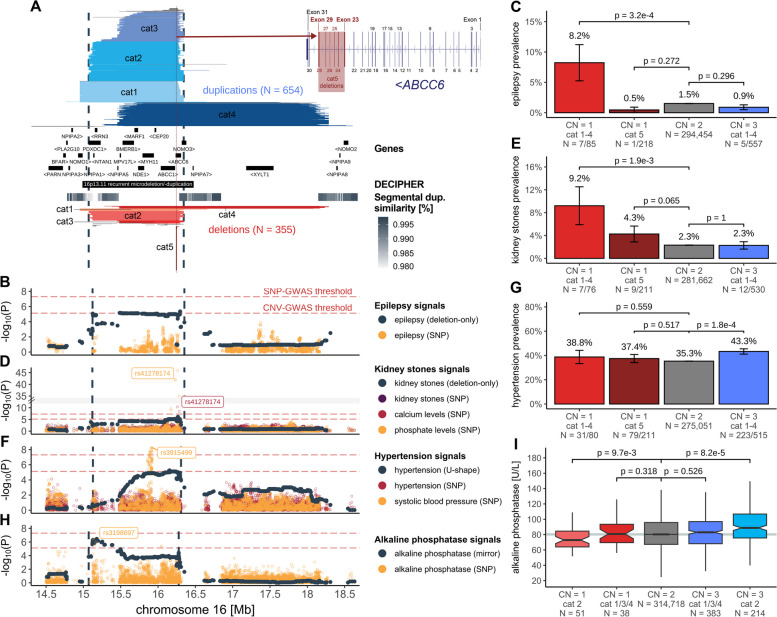
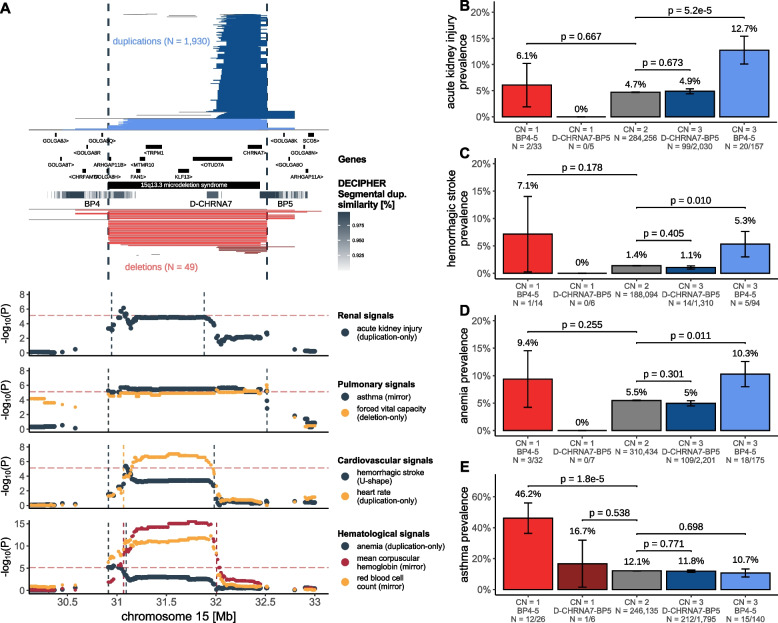
Results: We identified 73 signals involving 40 diseases, all of which indicating that CNVs increased disease risk and caused earlier onset. We estimated that 16% of these associations are indirect, acting by increasing body mass index (BMI). Signals mapped to 45 unique, non-overlapping regions, nine of which being linked to known GDs. Number and identity of genes affected by CNVs modulated their pathogenicity, with many associations being supported by colocalization with both common and rare single-nucleotide variant association signals. Dissection of association signals provided insights into the epidemiology of known gene-disease pairs (e.g., deletions in BRCA1 and LDLR increased risk for ovarian cancer and ischemic heart disease, respectively), clarified dosage mechanisms of action (e.g., both increased and decreased dosage of 17q12 impacted renal health), and identified putative causal genes (e.g., ABCC6 for kidney stones). Characterization of the pleiotropic pathological consequences of recurrent CNVs at 15q13, 16p13.11, 16p12.2, and 22q11.2 in adulthood indicated variable expressivity of these regions and the involvement of multiple genes. Finally, we show that while the total burden of rare CNVs-and especially deletions-strongly associated with disease risk, it only accounted for ~ 0.02% of the UKBB disease burden. These associations are mainly driven by CNVs at known GD CNV regions, whose pleiotropic effect on common diseases was broader than anticipated by our CNV-GWAS.
Conclusions: Our results shed light on the prominent role of rare CNVs in determining common disease susceptibility within the general population and provide actionable insights for anticipating later-onset comorbidities in carriers of recurrent CNVs.
Keywords: 16p11.2; 16p13.11; CNV; Common diseases; GWAS; Genomic disorders; Pleiotropy; Structural variation; Time-to-event analysis.
© 2023. The Author(s).
Conflict of interest statement
SO is an employee of MSD at the time of the submission; contribution to the research occurred during affiliation at the University of Lausanne. The remaining authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous