

MAGPIE: accurate pathogenic prediction for multiple variant types using machine learning approach
- PMID: 38185709
- PMCID: PMC10773112
- DOI: 10.1186/s13073-023-01274-4
MAGPIE: accurate pathogenic prediction for multiple variant types using machine learning approach
Erratum in
-
Correction: Genome Med 15, 115 & Genome Med 16, 3.Genome Med. 2024 May 14;16(1):68. doi: 10.1186/s13073-024-01343-2. Genome Med. 2024. PMID: 38745249 Free PMC article. No abstract available.
Abstract
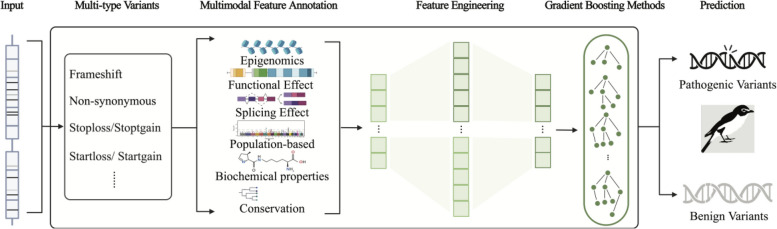
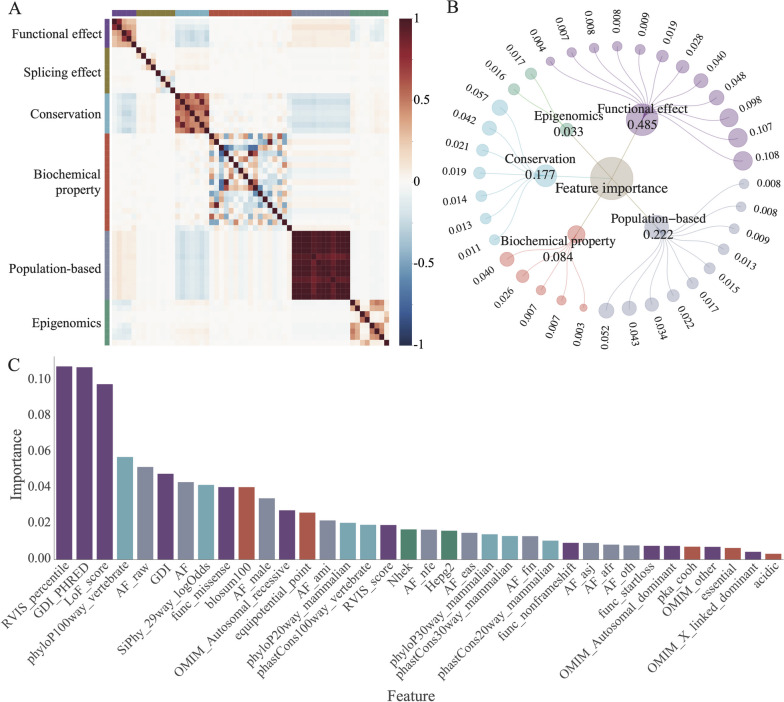
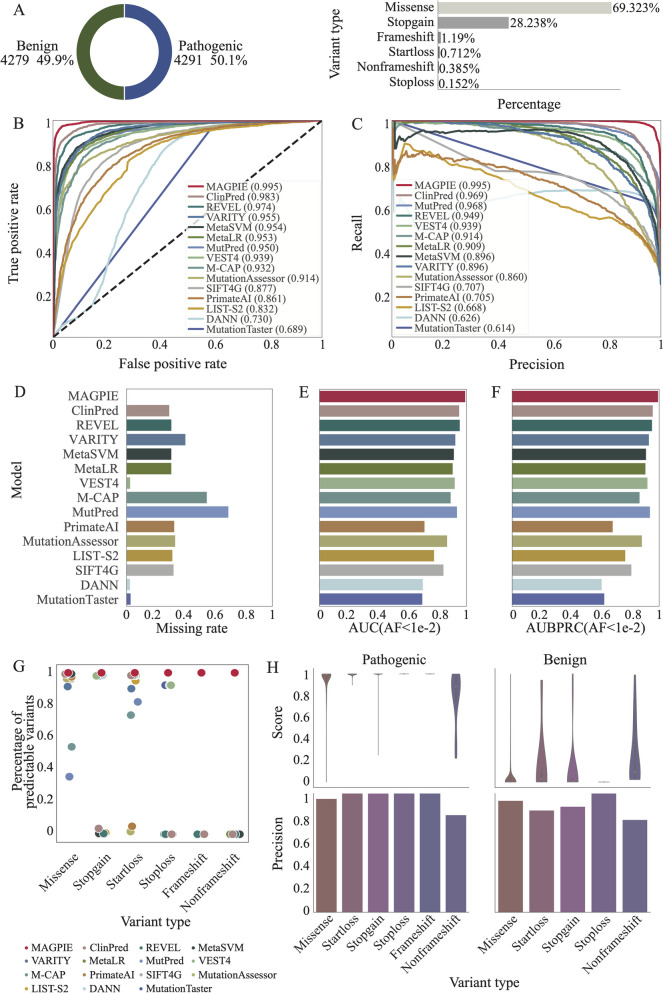
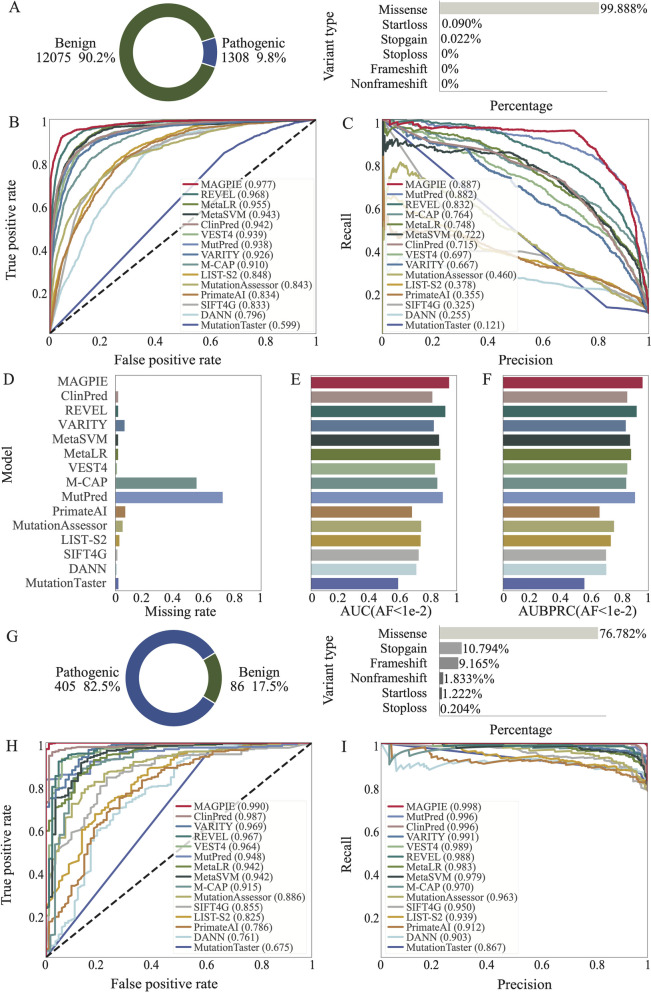
Identifying pathogenic variants from the vast majority of nucleotide variation remains a challenge. We present a method named Multimodal Annotation Generated Pathogenic Impact Evaluator (MAGPIE) that predicts the pathogenicity of multi-type variants. MAGPIE uses the ClinVar dataset for training and demonstrates superior performance in both the independent test set and multiple orthogonal validation datasets, accurately predicting variant pathogenicity. Notably, MAGPIE performs best in predicting the pathogenicity of rare variants and highly imbalanced datasets. Overall, results underline the robustness of MAGPIE as a valuable tool for predicting pathogenicity in various types of human genome variations. MAGPIE is available at https://github.com/shenlab-genomics/magpie .
Keywords: Genomic variation; Machine learning; Multimodal annotation; Pathogenic prediction.
© 2023. The Author(s).
Conflict of interest statement
The authors have submitted a patent application for the method. Other than this, the authors declare that they do not have any competing interests.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
