

This is a preprint.
AlphaFold2 structures template ligand discovery
- PMID: 38187536
- PMCID: PMC10769324
- DOI: 10.1101/2023.12.20.572662
AlphaFold2 structures template ligand discovery
Update in
-
AlphaFold2 structures guide prospective ligand discovery.Science. 2024 Jun 21;384(6702):eadn6354. doi: 10.1126/science.adn6354. Epub 2024 Jun 21. Science. 2024. PMID: 38753765 Free PMC article.
Abstract
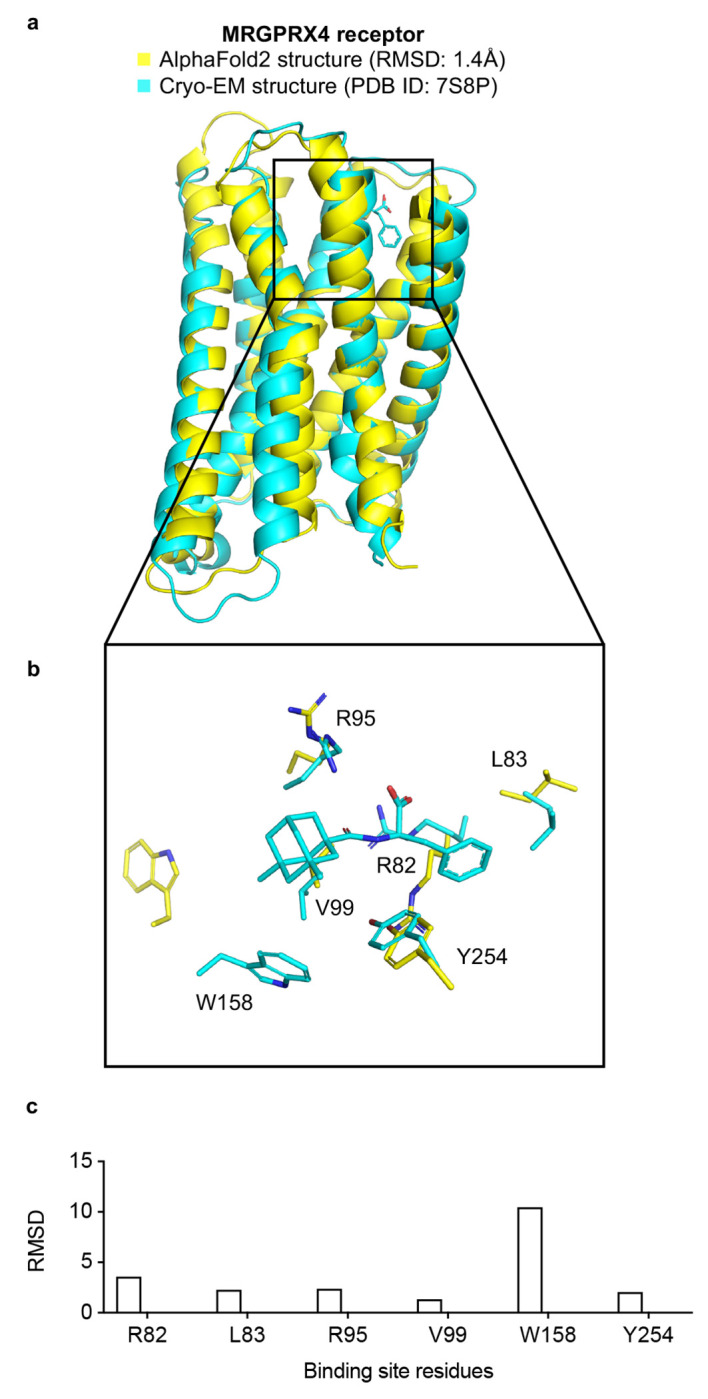
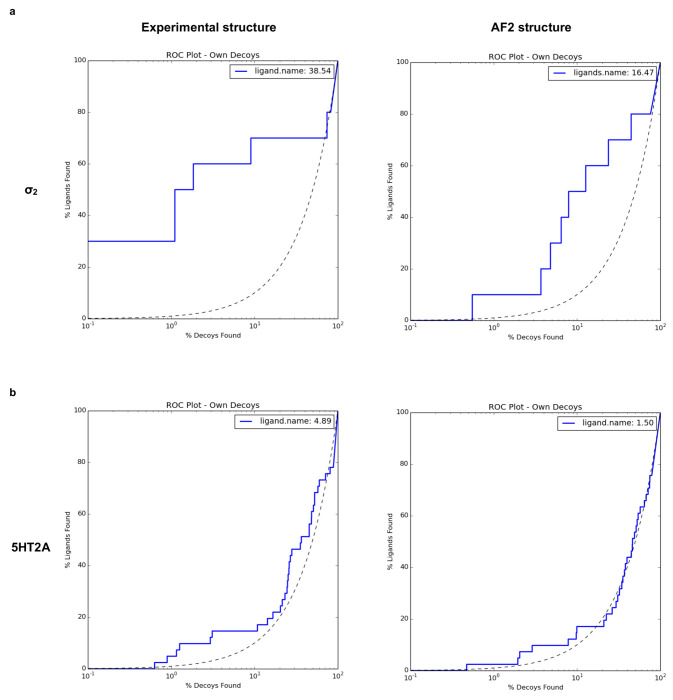
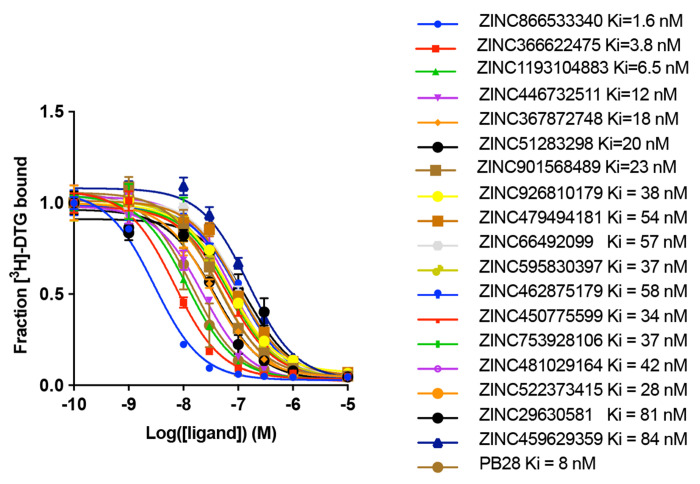
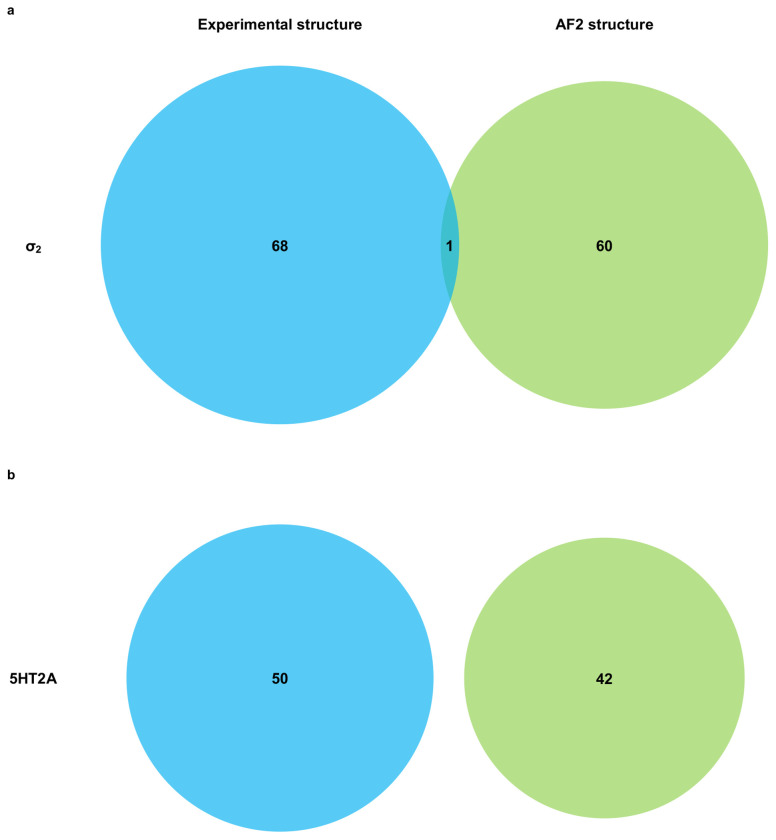
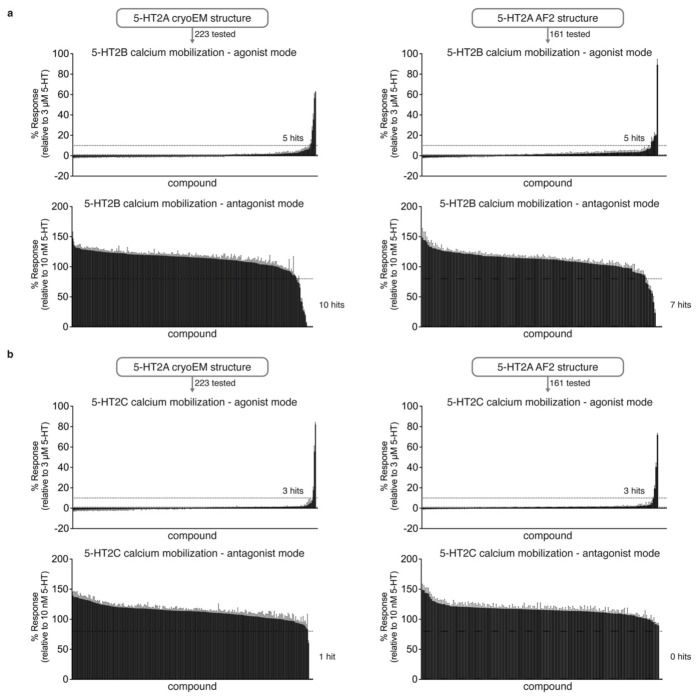
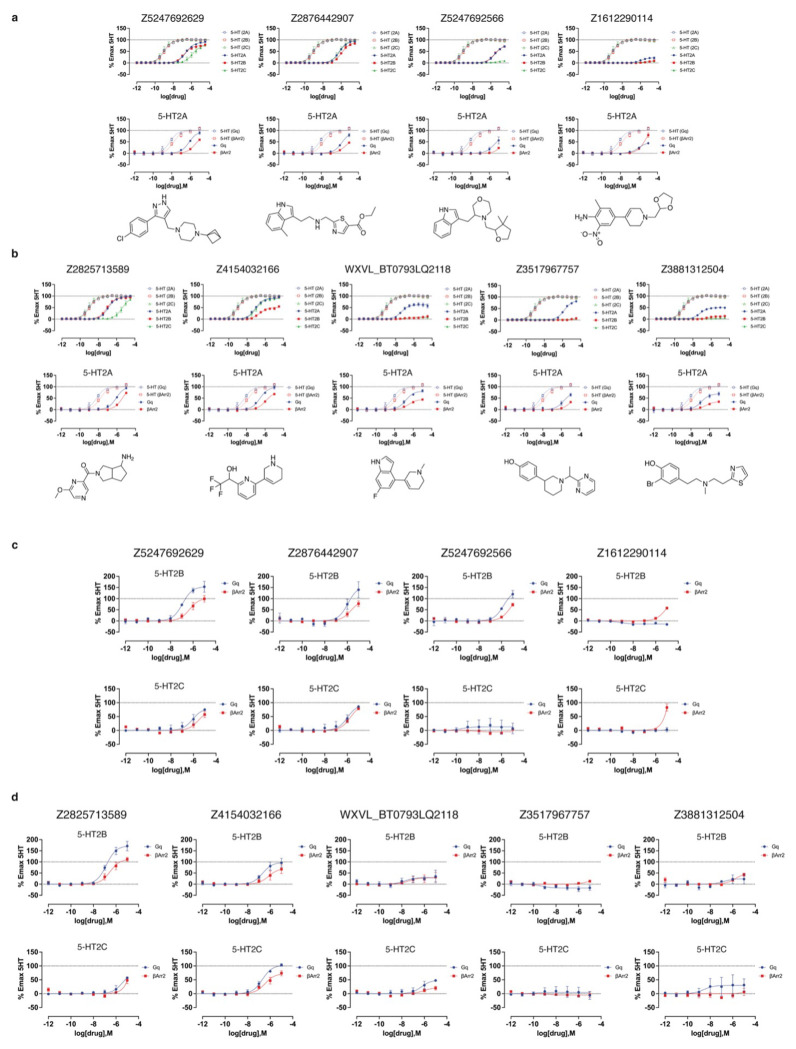
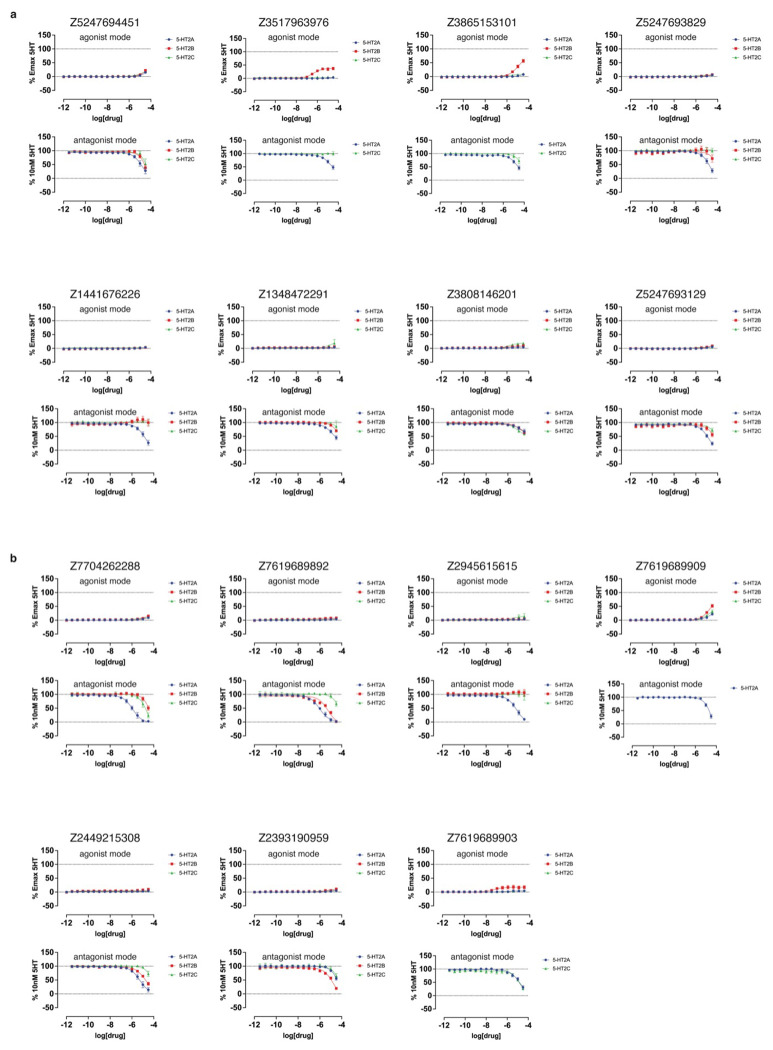
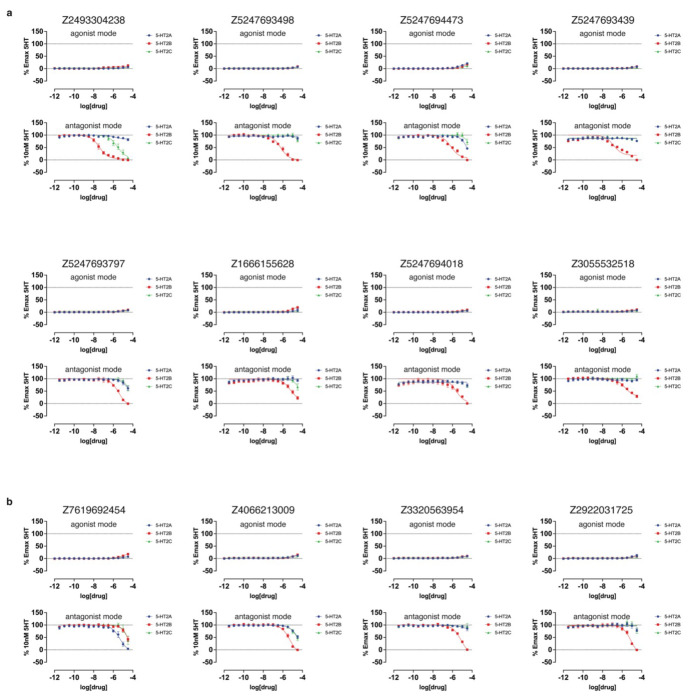
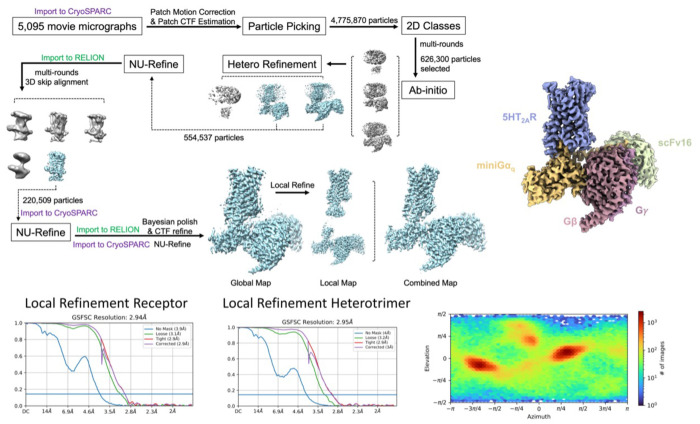
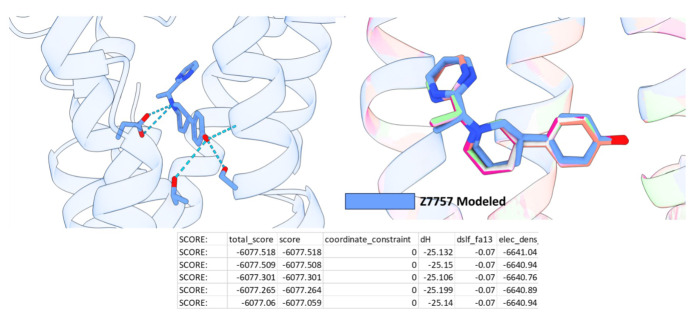
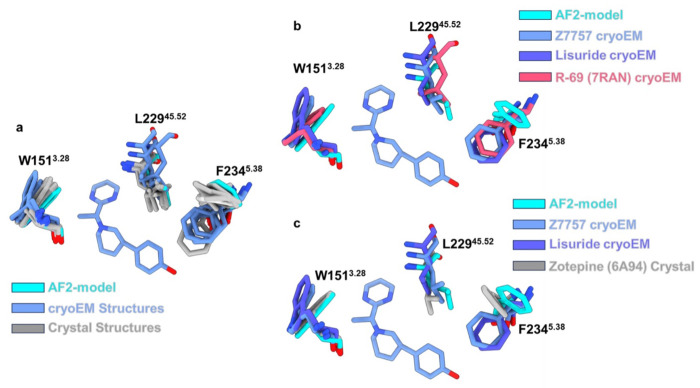
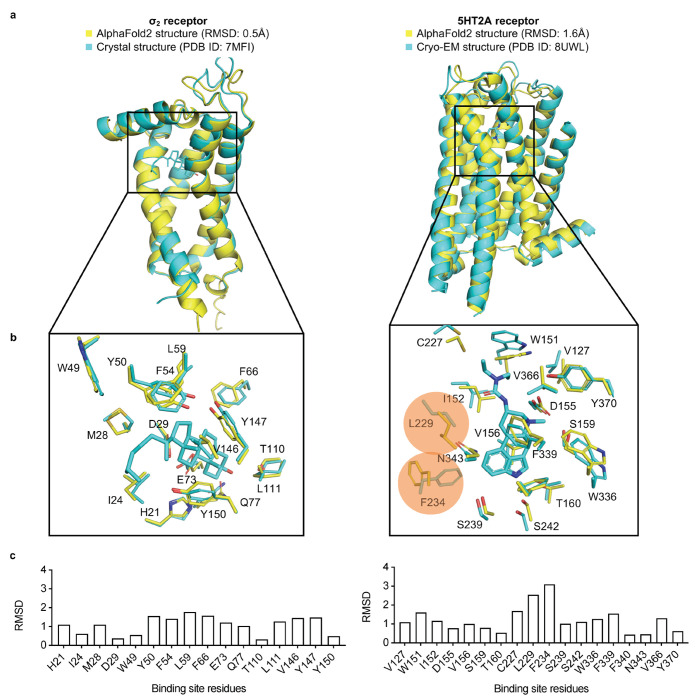
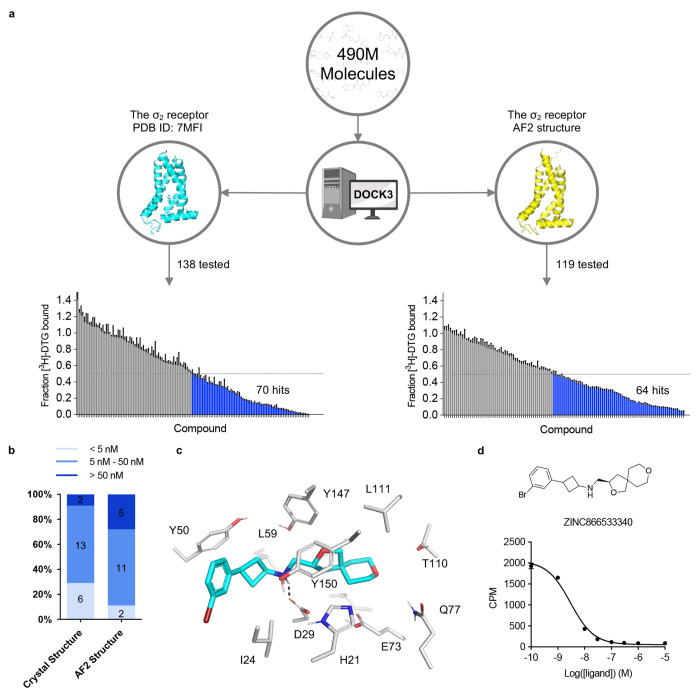
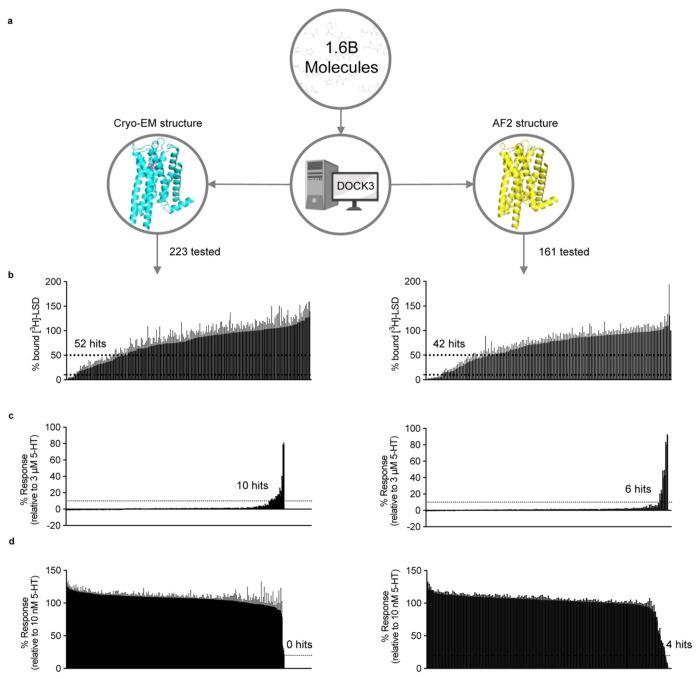
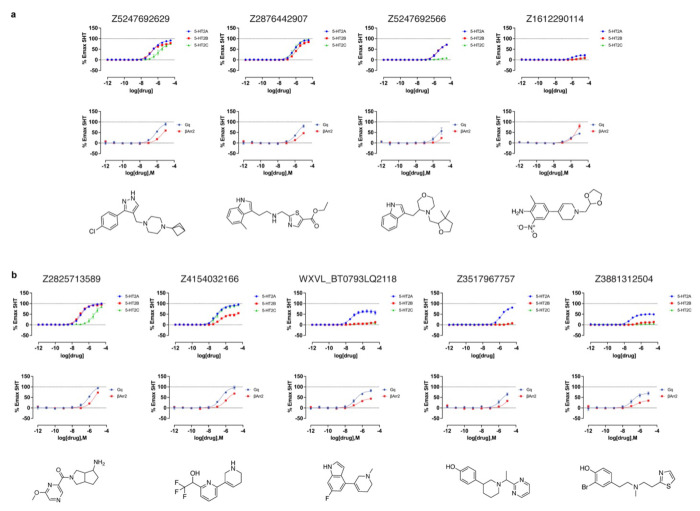
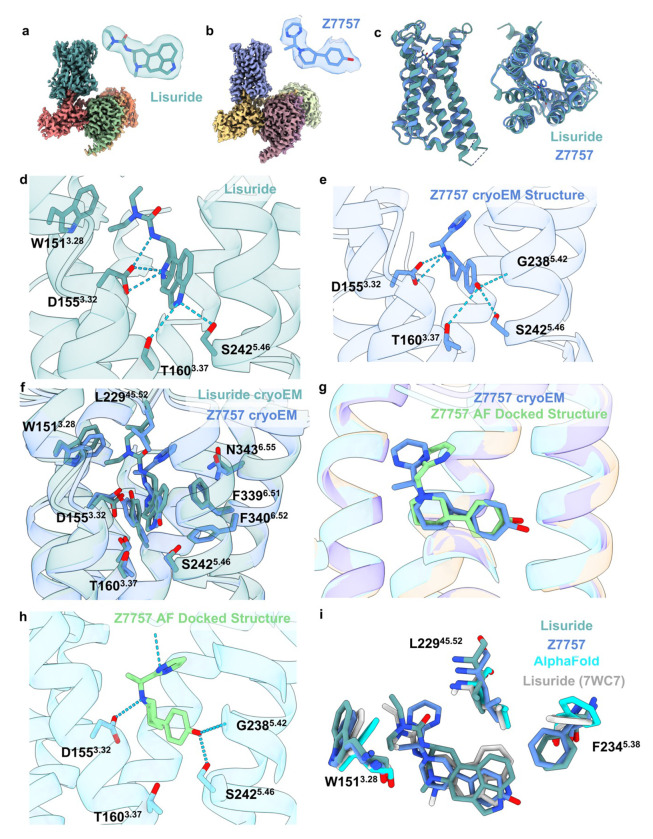
AlphaFold2 (AF2) and RosettaFold have greatly expanded the number of structures available for structure-based ligand discovery, even though retrospective studies have cast doubt on their direct usefulness for that goal. Here, we tested unrefined AF2 models prospectively, comparing experimental hit-rates and affinities from large library docking against AF2 models vs the same screens targeting experimental structures of the same receptors. In retrospective docking screens against the σ2 and the 5-HT2A receptors, the AF2 structures struggled to recapitulate ligands that we had previously found docking against the receptors' experimental structures, consistent with published results. Prospective large library docking against the AF2 models, however, yielded similar hit rates for both receptors versus docking against experimentally-derived structures; hundreds of molecules were prioritized and tested against each model and each structure of each receptor. The success of the AF2 models was achieved despite differences in orthosteric pocket residue conformations for both targets versus the experimental structures. Intriguingly, against the 5-HT2A receptor the most potent, subtype-selective agonists were discovered via docking against the AF2 model, not the experimental structure. To understand this from a molecular perspective, a cryoEM structure was determined for one of the more potent and selective ligands to emerge from docking against the AF2 model of the 5-HT2A receptor. Our findings suggest that AF2 models may sample conformations that are relevant for ligand discovery, much extending the domain of applicability of structure-based ligand discovery.
Conflict of interest statement
Competing interests B.K.S. is co-founder of BlueDolphin, LLC, Epiodyne, and Deep Apple Therapeutics, Inc., serves on the SRB of Genentech, the SAB of Schrodinger LLC and of Vilya Therapeutics, and consults for Levator Therapeutics, Hyku Therapeutics, and for Great Point Ventures. J.J.I. cofounded Deep Apple Therapeutics, Inc., and BlueDolphin, LLC. B.L.R is a co-founder of Epiodyne and Onsero and on the SAB for Onsero, Epiodyne, Levator, Escient and Septerna. A.C.K. is a cofounder and consultant for biotechnology companies Tectonic Therapeutic and Seismic Therapeutic, and also for the Institute for Protein Innovation, a nonprofit research institute. X.B.A is now a senior scientist at Tectonic Therapeutics
Figures
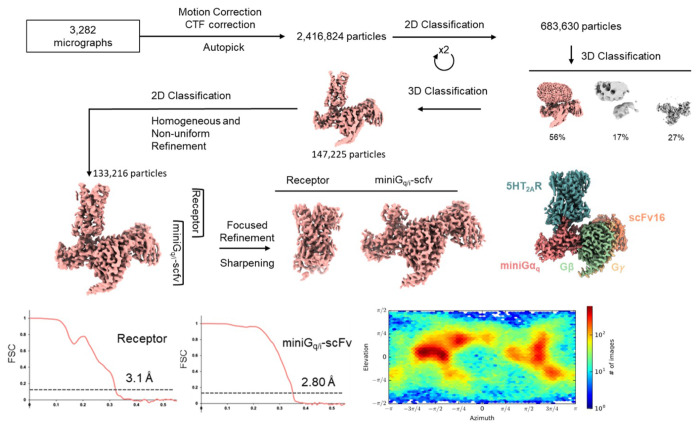
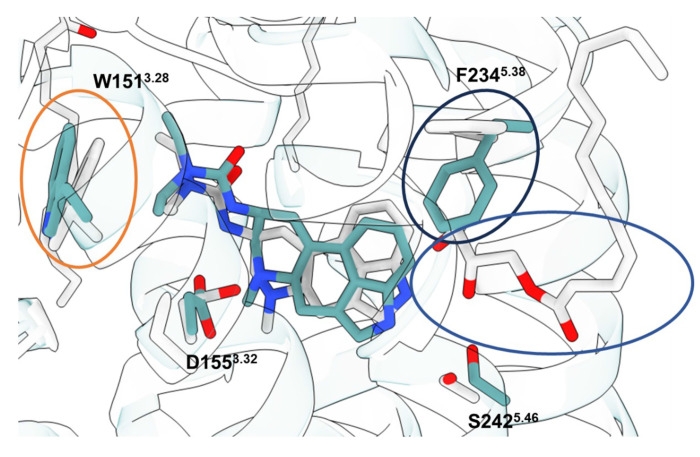
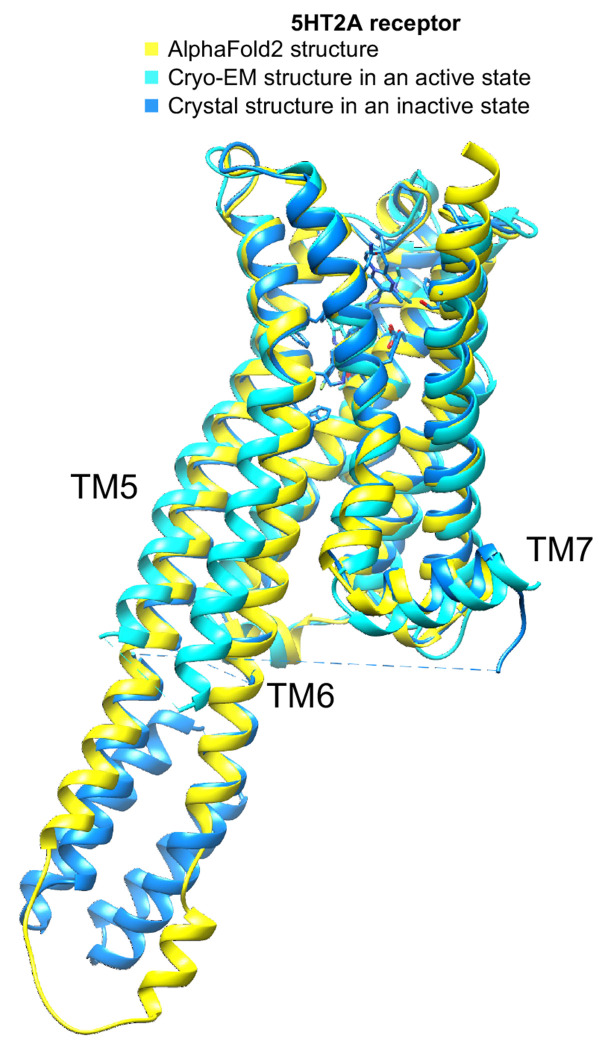
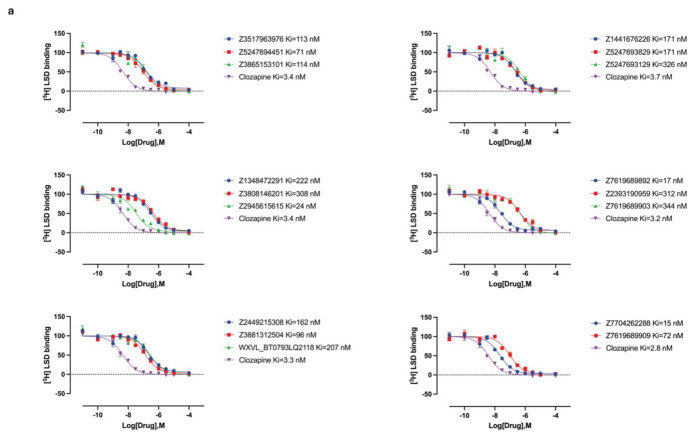
References
-
- Sadybekov A. V. & Katritch V. Computational approaches streamlining drug discovery. Nature 616, 673–685 (2023). - PubMed
-
- Wang Y. et al. Structures of the entire human opioid receptor family. Cell 186, 413–427. e417 (2023). - PubMed
-
- Xu P. et al. Structural insights into the lipid and ligand regulation of serotonin receptors. Nature 592, 469–473 (2021). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous