

This is a preprint.
Rewriting regulatory DNA to dissect and reprogram gene expression
- PMID: 38187584
- PMCID: PMC10769263
- DOI: 10.1101/2023.12.20.572268
Rewriting regulatory DNA to dissect and reprogram gene expression
Update in
-
Rewriting regulatory DNA to dissect and reprogram gene expression.Cell. 2025 Jun 12;188(12):3349-3366.e23. doi: 10.1016/j.cell.2025.03.034. Epub 2025 Apr 16. Cell. 2025. PMID: 40245860
Abstract
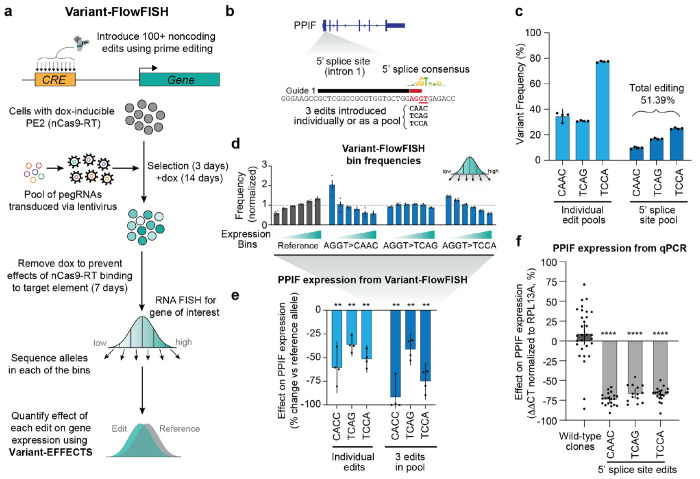
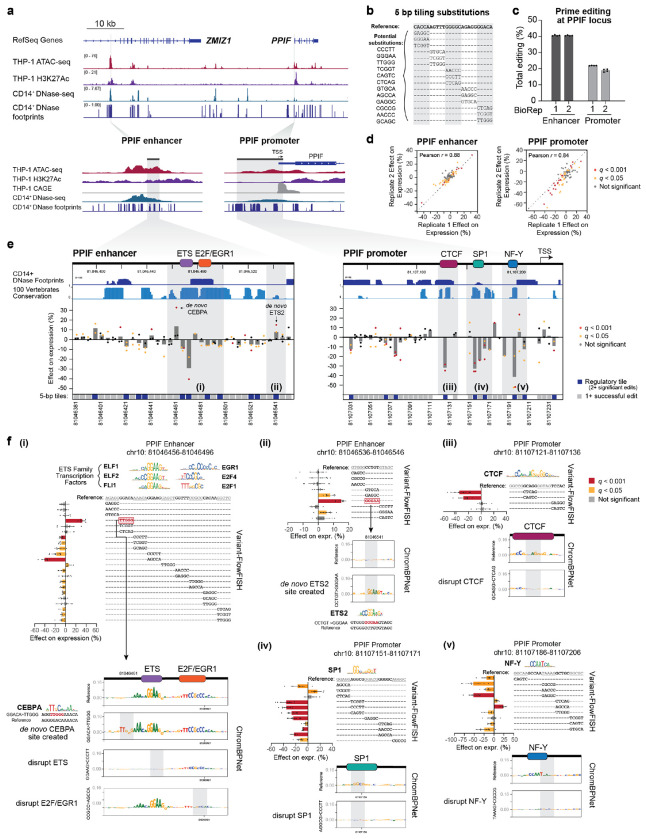
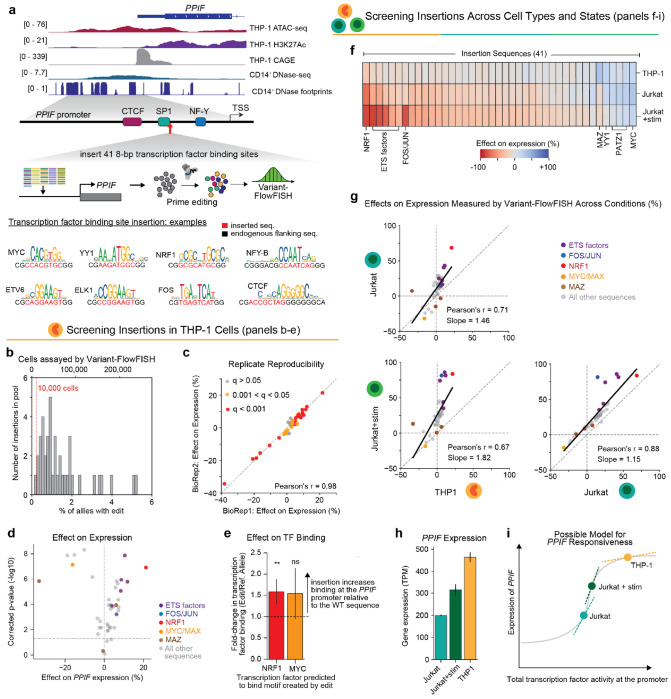
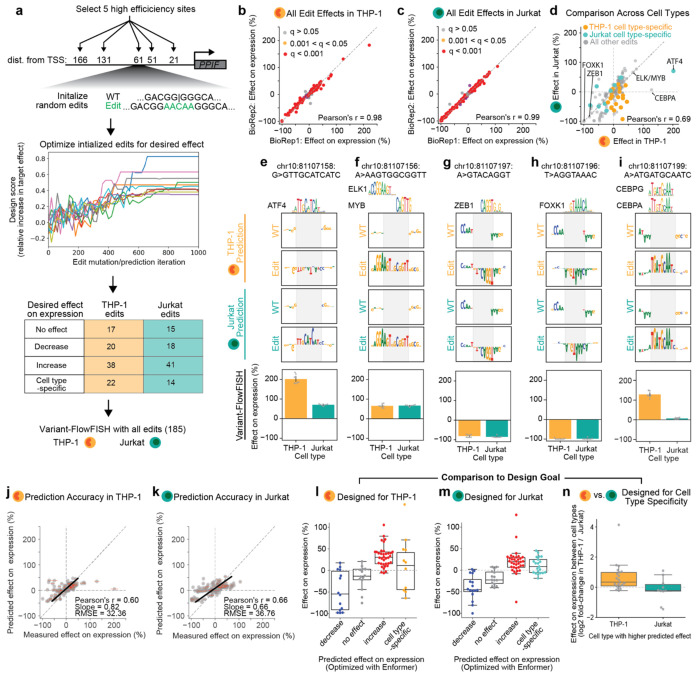
Regulatory DNA sequences within enhancers and promoters bind transcription factors to encode cell type-specific patterns of gene expression. However, the regulatory effects and programmability of such DNA sequences remain difficult to map or predict because we have lacked scalable methods to precisely edit regulatory DNA and quantify the effects in an endogenous genomic context. Here we present an approach to measure the quantitative effects of hundreds of designed DNA sequence variants on gene expression, by combining pooled CRISPR prime editing with RNA fluorescence in situ hybridization and cell sorting (Variant-FlowFISH). We apply this method to mutagenize and rewrite regulatory DNA sequences in an enhancer and the promoter of PPIF in two immune cell lines. Of 672 variant-cell type pairs, we identify 497 that affect PPIF expression. These variants appear to act through a variety of mechanisms including disruption or optimization of existing transcription factor binding sites, as well as creation of de novo sites. Disrupting a single endogenous transcription factor binding site often led to large changes in expression (up to -40% in the enhancer, and -50% in the promoter). The same variant often had different effects across cell types and states, demonstrating a highly tunable regulatory landscape. We use these data to benchmark performance of sequence-based predictive models of gene regulation, and find that certain types of variants are not accurately predicted by existing models. Finally, we computationally design 185 small sequence variants (≤10 bp) and optimize them for specific effects on expression in silico. 84% of these rationally designed edits showed the intended direction of effect, and some had dramatic effects on expression (-100% to +202%). Variant-FlowFISH thus provides a powerful tool to map the effects of variants and transcription factor binding sites on gene expression, test and improve computational models of gene regulation, and reprogram regulatory DNA.
Conflict of interest statement
Competing Interests J.M.E. is a consultant and equity holder in Martingale Labs, Inc., has received materials from 10x Genomics unrelated to this study, and has received speaking honoraria from GSK plc. J.L. and D.R.K. are employed by Calico Life Sciences LLC. C.P.F. is employed by Sanofi. A.K. is on the scientific advisory board of PatchBio, SerImmune and OpenTargets, was a consultant with Illumina, and owns shares in DeepGenomics, ImmunAI and Freenome. M.T.M., G.E.M., B.R.D., H.J., K.G., and J.M.E. are inventors on a provisional patent application related to this work.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials