

This is a preprint.
DNA-sequence and epigenomic determinants of local rates of transcription elongation
- PMID: 38187771
- PMCID: PMC10769381
- DOI: 10.1101/2023.12.21.572932
DNA-sequence and epigenomic determinants of local rates of transcription elongation
Abstract
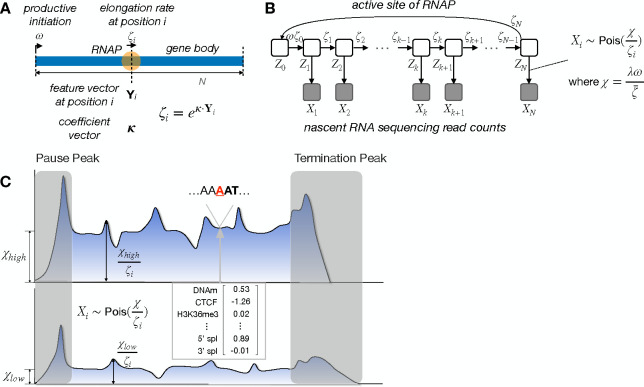
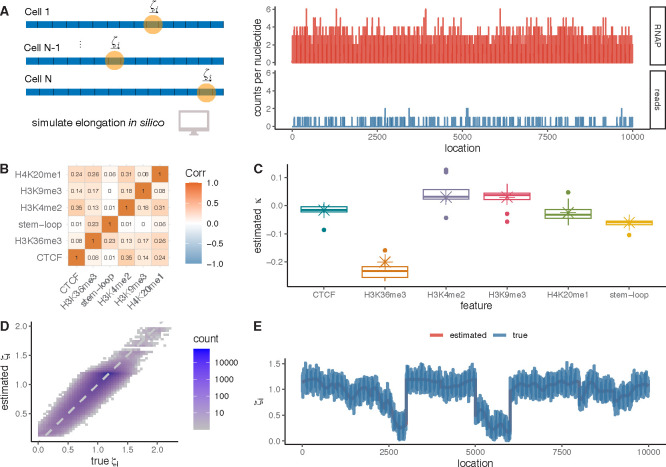
Across all branches of life, transcription elongation is a crucial, regulated phase in gene expression. Many recent studies in eukaryotes have focused on the regulation of promoter-proximal pausing of RNA Polymerase II (Pol II), but rates of productive elongation also vary substantially throughout the gene body, both within and across genes. Here, we introduce a probabilistic model for systematically evaluating potential determinants of the local elongation rate based on nascent RNA sequencing (NRS) data. Our model is derived from a unified model for both the kinetics of Pol II movement along the DNA template and the generation of NRS read counts at steady state. It allows for a continuously variable elongation rate along the gene body, with the rate at each nucleotide defined by a generalized linear relationship with nearby genomic and epigenomic features. High-dimensional feature vectors are accommodated through a sparse-regression extension. We show with simulations that the model allows accurate detection of associated features and accurate prediction of local elongation rates. In an analysis of public PRO-seq and epigenomic data, we identify several features that are strongly associated with reductions in the local elongation rate, including DNA methylation, splice sites, RNA stem-loops, CTCF binding sites, and several histone marks, including H3K36me3 and H4K20me1. By contrast, low-complexity sequences and H3K79me2 marks are associated with increases in elongation rate. In an analysis of DNA -mers, we find that cytosine nucleotides are strongly associated with reductions in local elongation rate, particularly when preceded by guanines and followed by adenines or thymines. Increases in elongation rate are associated with thymines and A+T-rich -mers. These associations are generally shared across cell types, and by considering them our model is effective at predicting features of held-out PRO-seq data. Overall, our analysis is the first to permit genome-wide predictions of relative nucleotide-specific elongation rates based on complex sets of genomic and epigenomic covariates. We have made predictions available for the K562, CD14+, MCF-7, and HeLa-S3 cell types in a UCSC Genome Browser track.
Conflict of interest statement
Conflict of Interest The authors declare no competing interests.
Figures




References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials