

Modulations in the host cell proteome by the hantavirus nucleocapsid protein
- PMID: 38190410
- PMCID: PMC10798635
- DOI: 10.1371/journal.ppat.1011925
Modulations in the host cell proteome by the hantavirus nucleocapsid protein
Abstract
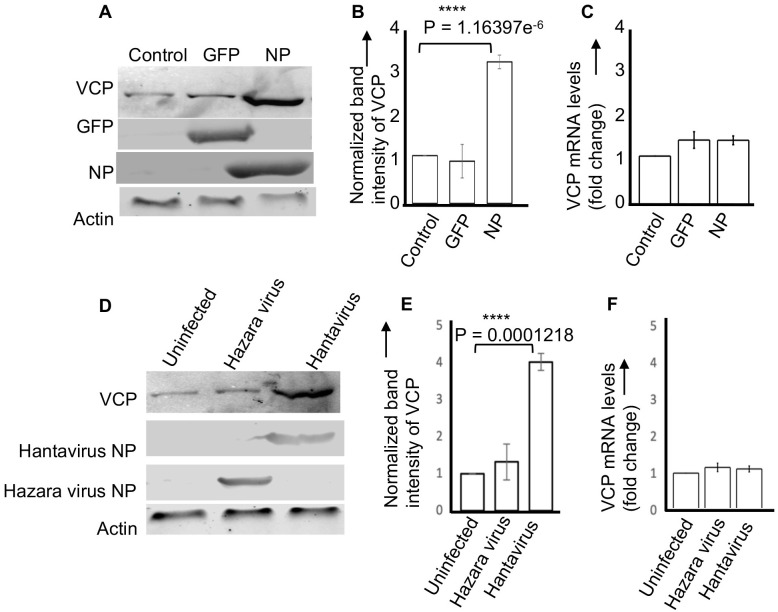
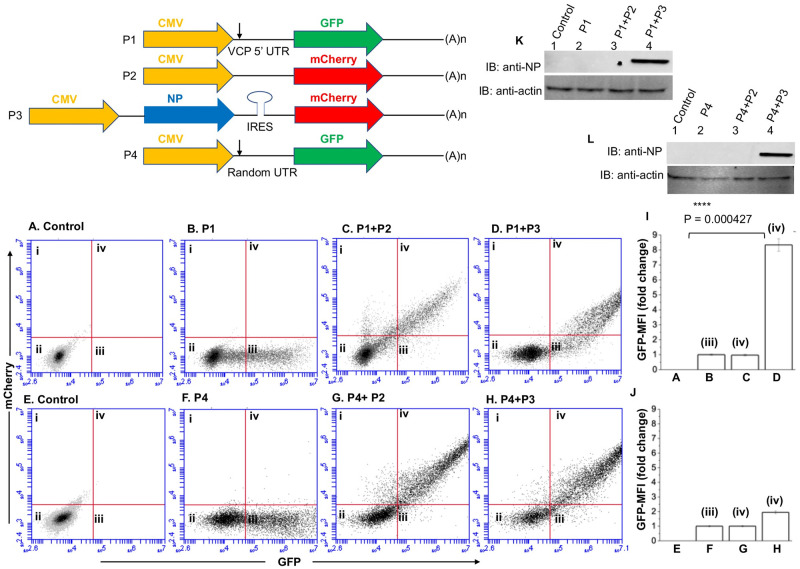
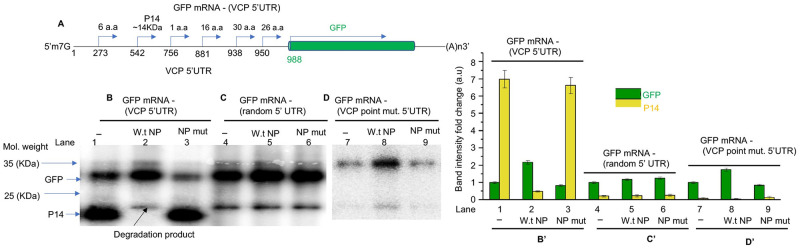
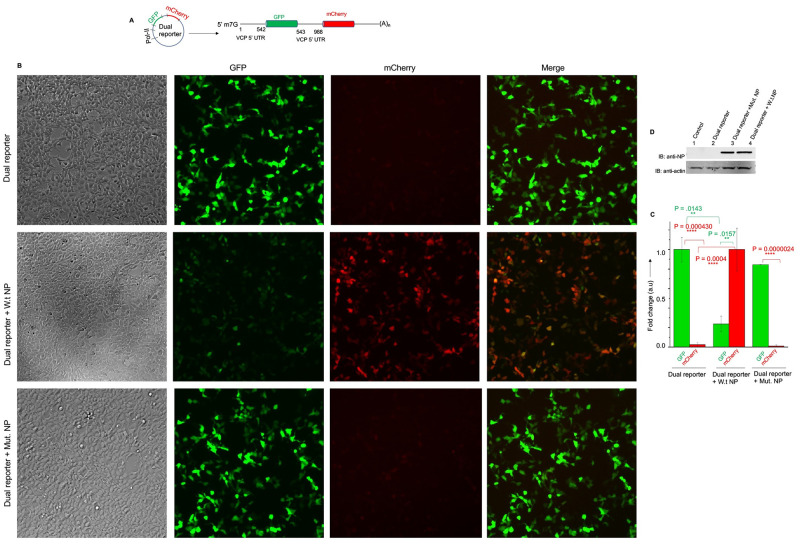
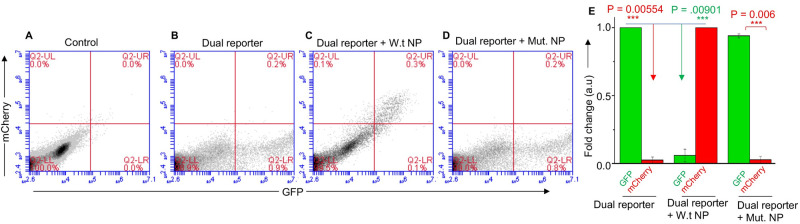
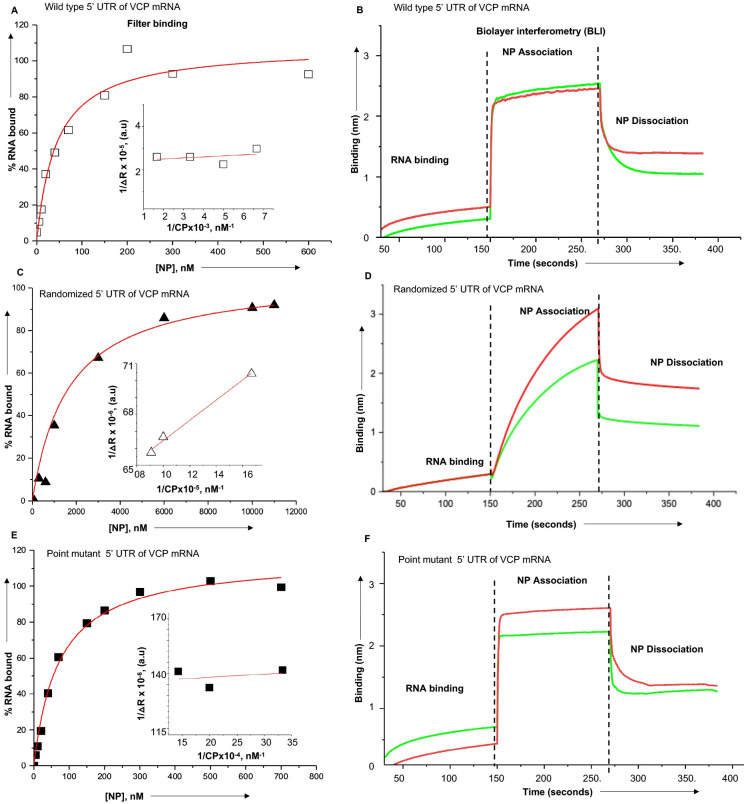
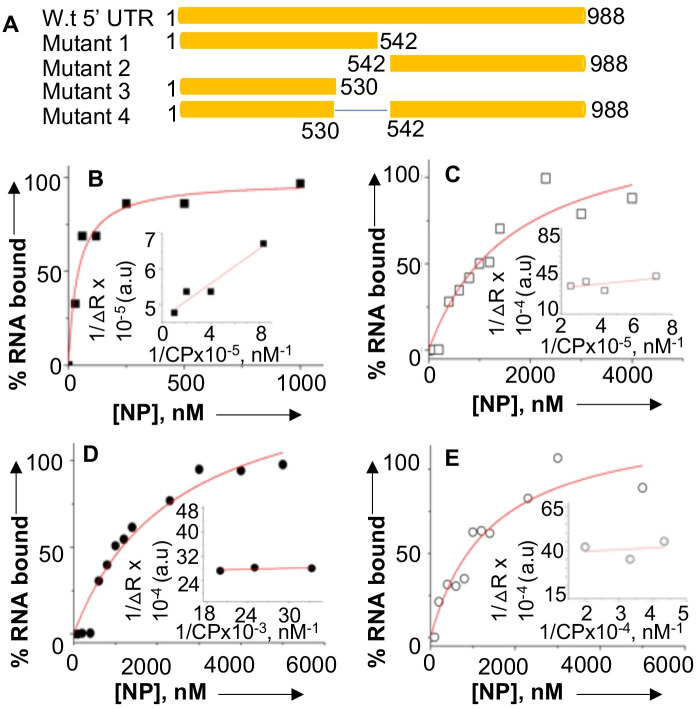
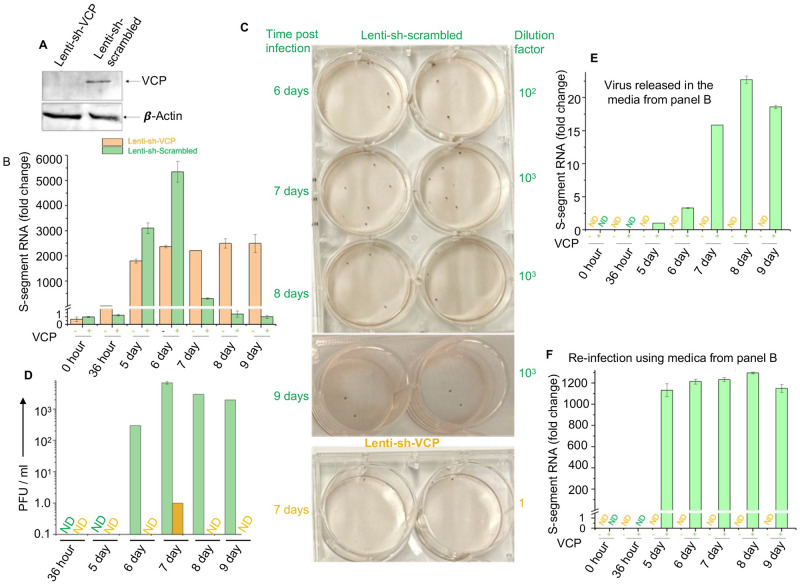
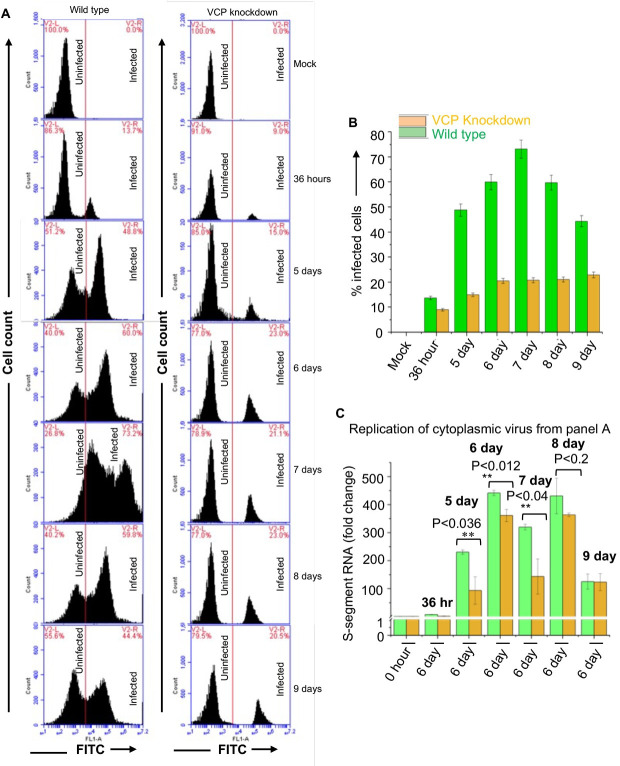
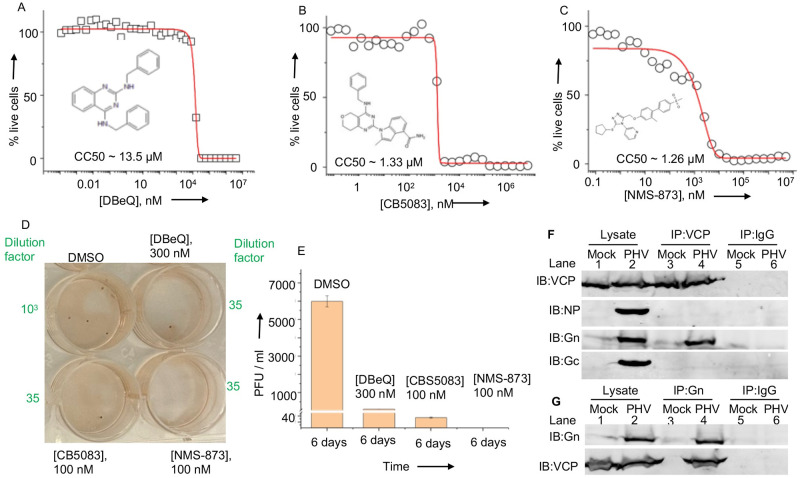
Hantaviruses have evolved a unique translation strategy to boost the translation of viral mRNA in infected cells. Hantavirus nucleocapsid protein (NP) binds to the viral mRNA 5' UTR and the 40S ribosomal subunit via the ribosomal protein S19. NP associated ribosomes are selectively loaded on viral transcripts to boost their translation. Here we demonstrate that NP expression upregulated the steady-state levels of a subset of host cell factors primarily involved in protein processing in the endoplasmic reticulum. Detailed investigation of Valosin-containing protein (VCP/p97), one of the upregulated host factors, in both transfected and virus infected cells revealed that NP with the assistance of VCP mRNA 5' UTR facilitates the translation of downstream VCP ORF. The VCP mRNA contains a 5' UTR of 987 nucleotides harboring six unusual start codons upstream of the correct start codon for VCP which is located at 988th position from the 5' cap. In vitro translation of a GFP reporter transcript harboring the VCP mRNA 5' UTR generated both GFP and a short polypeptide of ~14 KDa by translation initiation from start codon located in the 5' UTR at 542nd position from the 5' cap. The translation initiation from 542nd AUG in the UTR sequence was confirmed in cells using a dual reporter construct expressing mCherry and GFP. The synthesis of 14KDa polypeptide dramatically inhibited the translation of the ORF from the downstream correct start codon at 988th position from the 5' cap. We report that purified NP binds to the VCP mRNA 5' UTR with high affinity and NP binding site is located close to the 542ndAUG. NP binding shuts down the translation of 14KDa polypeptide which then facilitates the translation initiation at the correct AUG codon. Knockdown of VCP generated lower levels of poorly infectious hantavirus particle in the cellular cytoplasm whose egress was dramatically inhibited in human umbilical vein endothelial cells. We demonstrated that VCP binds to the hantavirus glycoprotein Gn before its incorporation into assembled virions and facilitates viral spread to neighboring cells during infection. Our results suggest that ribosome engagement at the 542nd AUG codon in the 5' UTR likely regulates the endogenous steady state levels of VCP in cells. Hantaviruses interrupt this regulatory mechanism to enhance the steady state levels of VCP in virus infected cells. This augmentation facilitates virus replication, supports the transmission of the virus to adjacent cells, and promotes the release of infectious virus particles from the host cell.
Copyright: © 2024 Royster et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
