

A pairwise cytokine code explains the organism-wide response to sepsis
- PMID: 38191855
- PMCID: PMC10834370
- DOI: 10.1038/s41590-023-01722-8
A pairwise cytokine code explains the organism-wide response to sepsis
Abstract
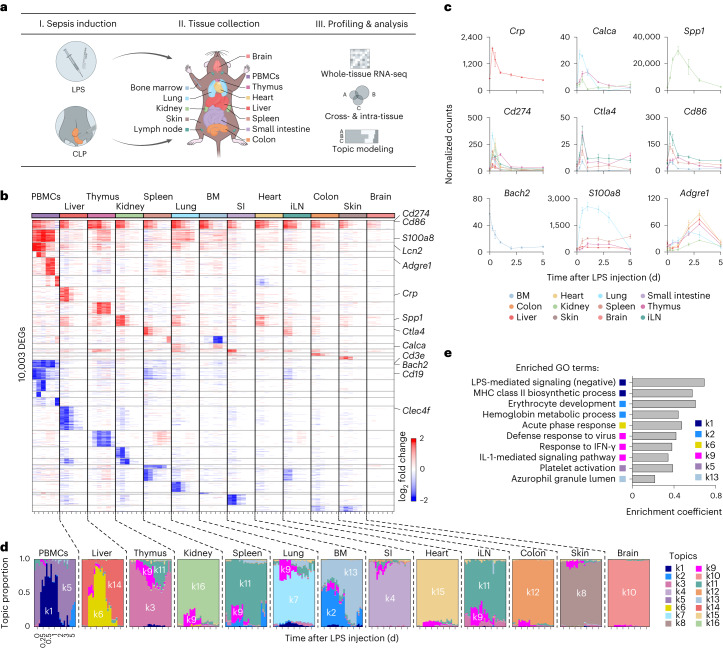
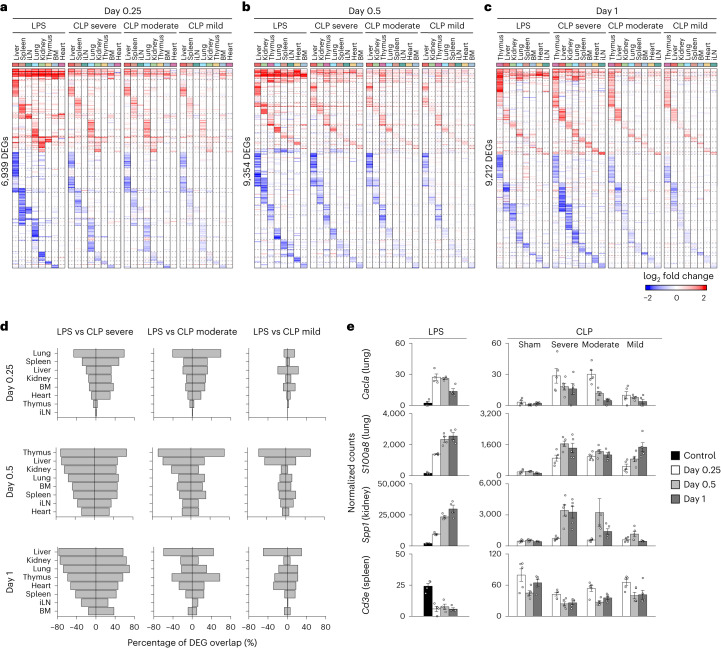
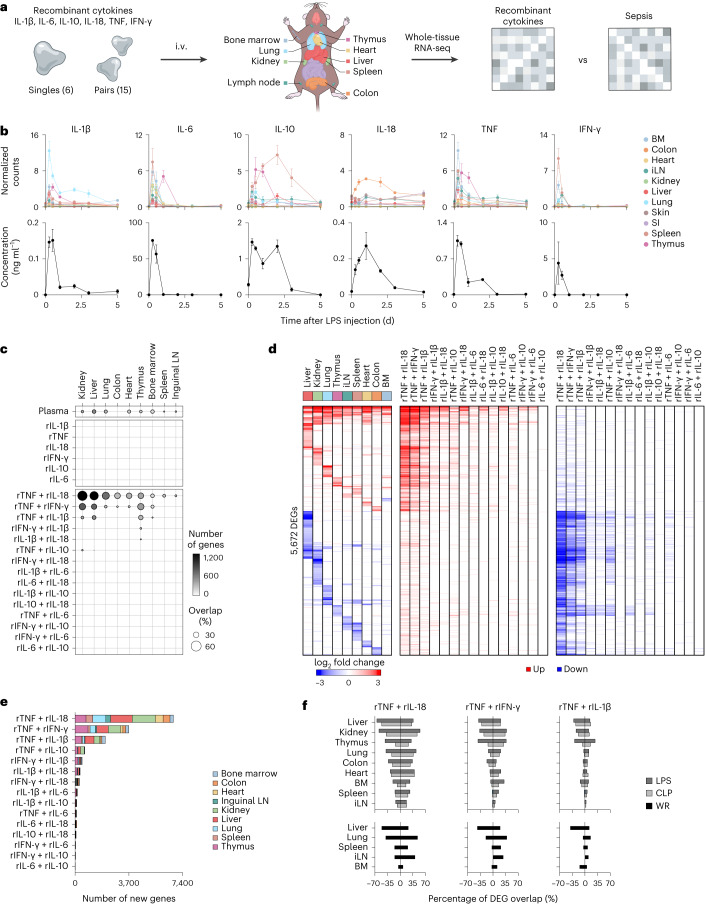
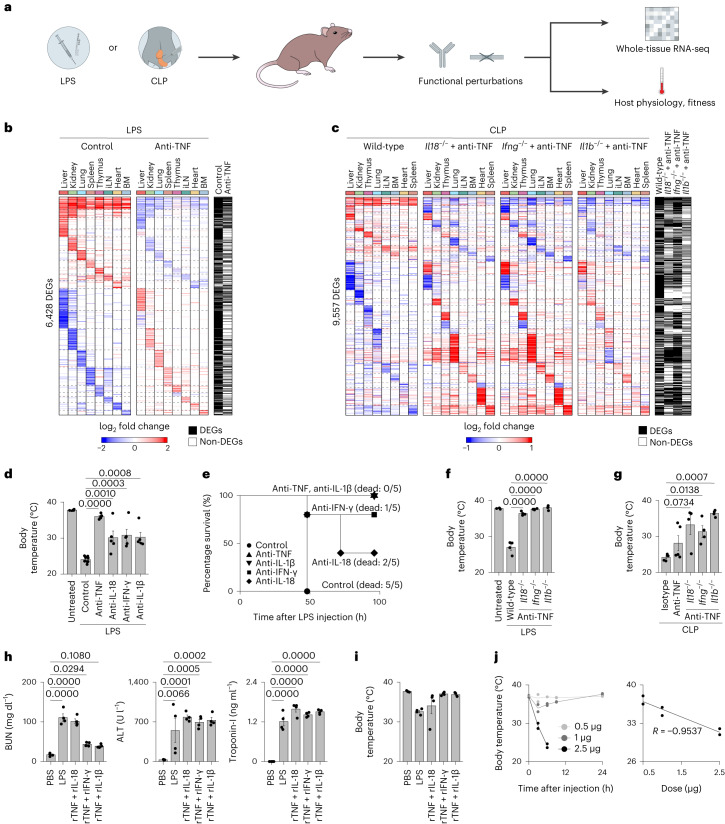
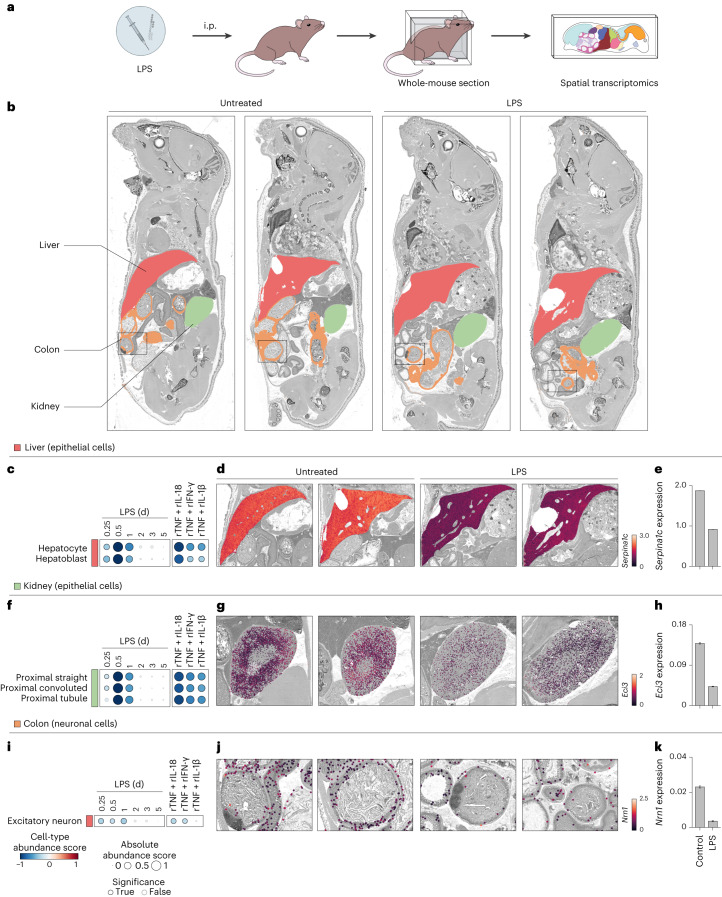
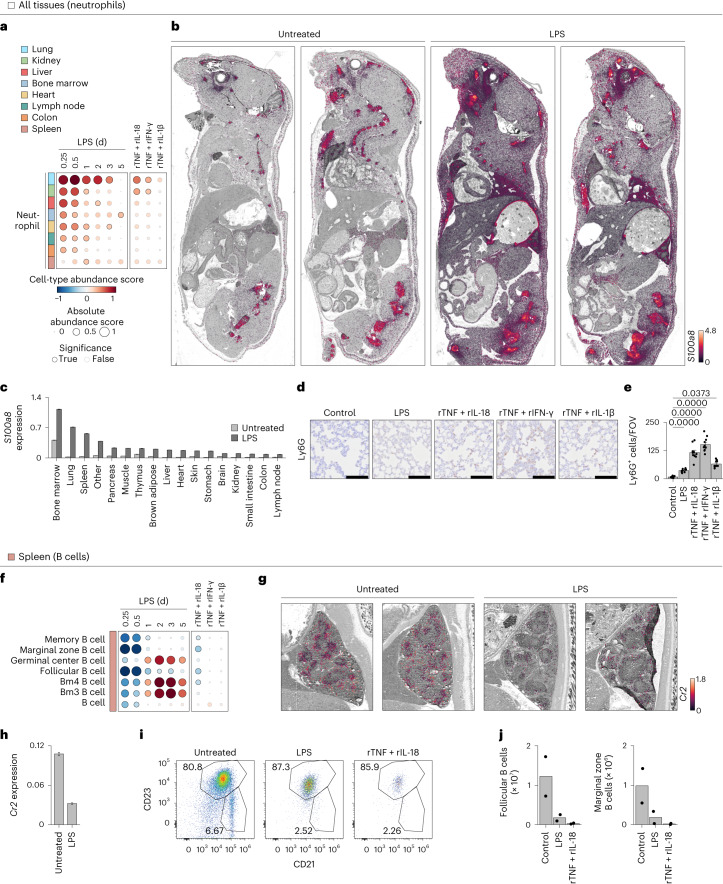
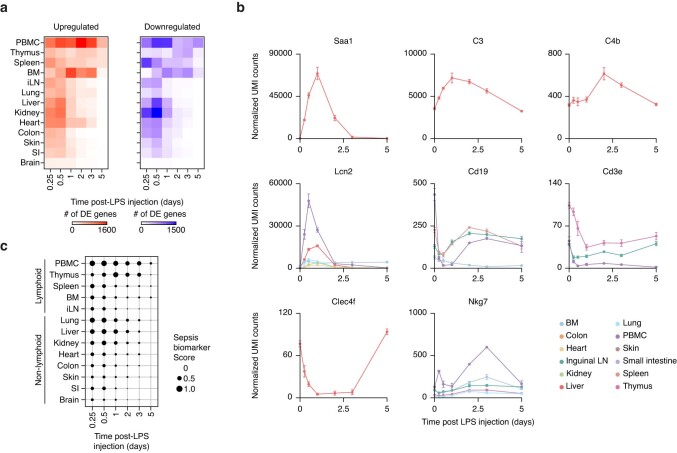
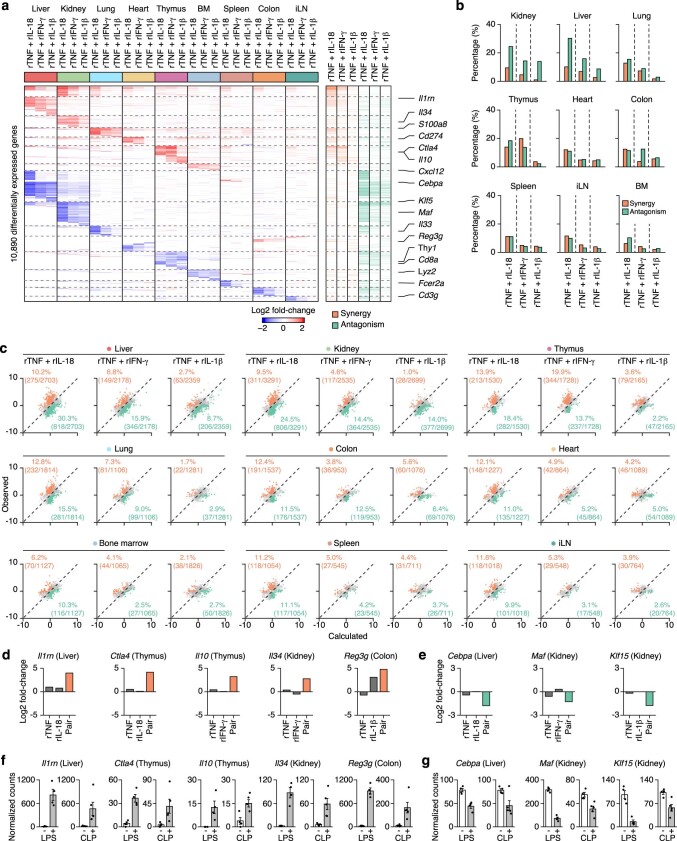
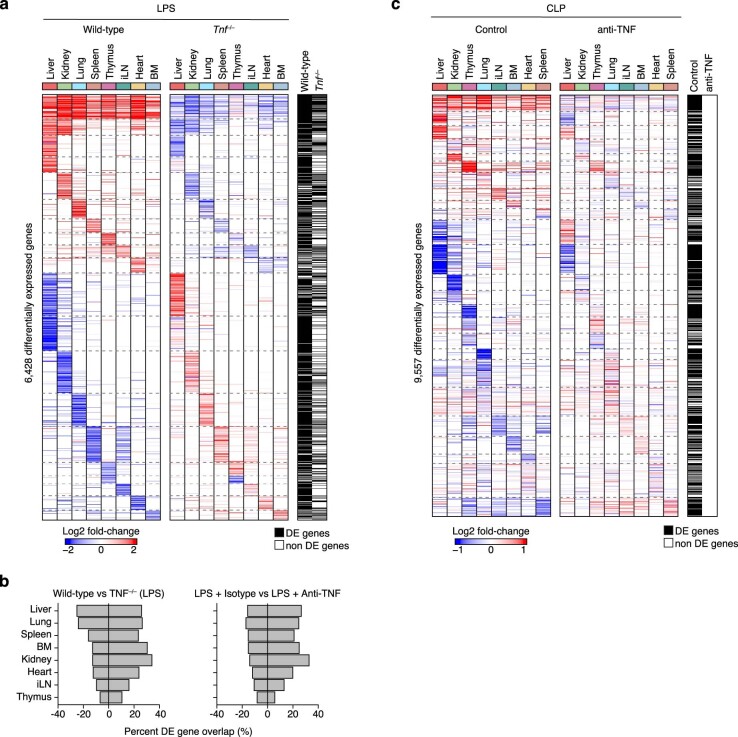
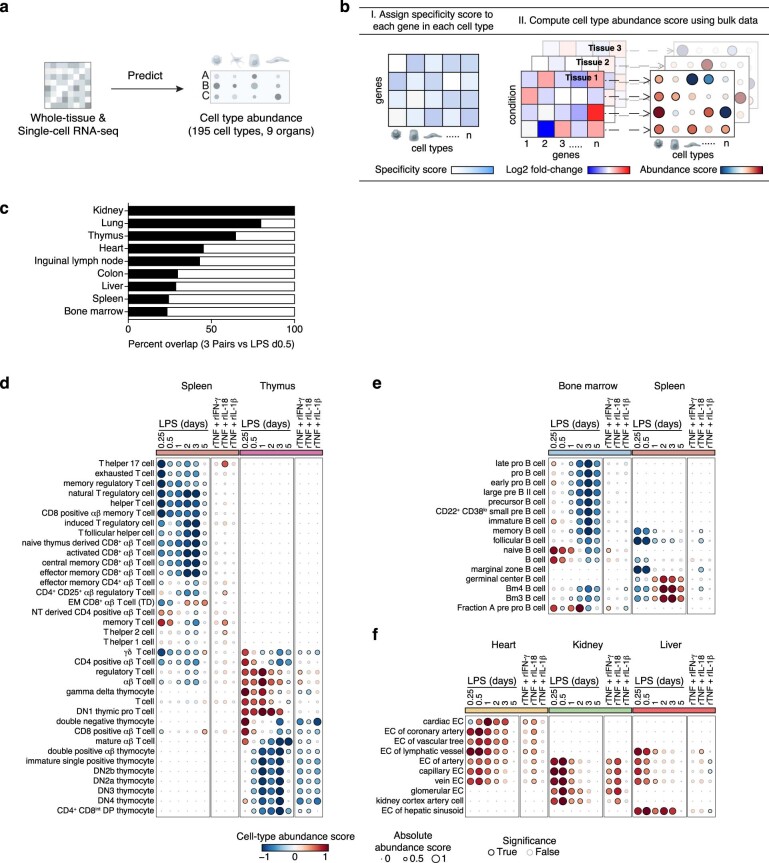
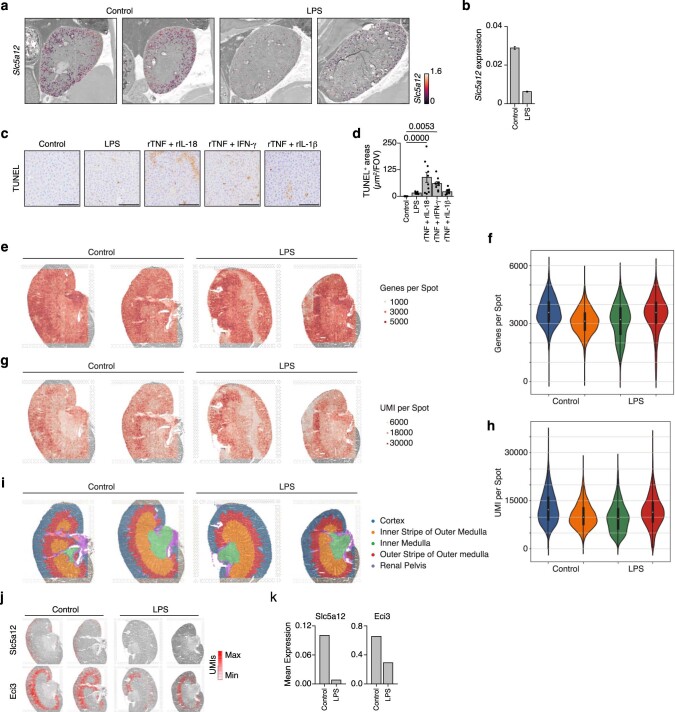
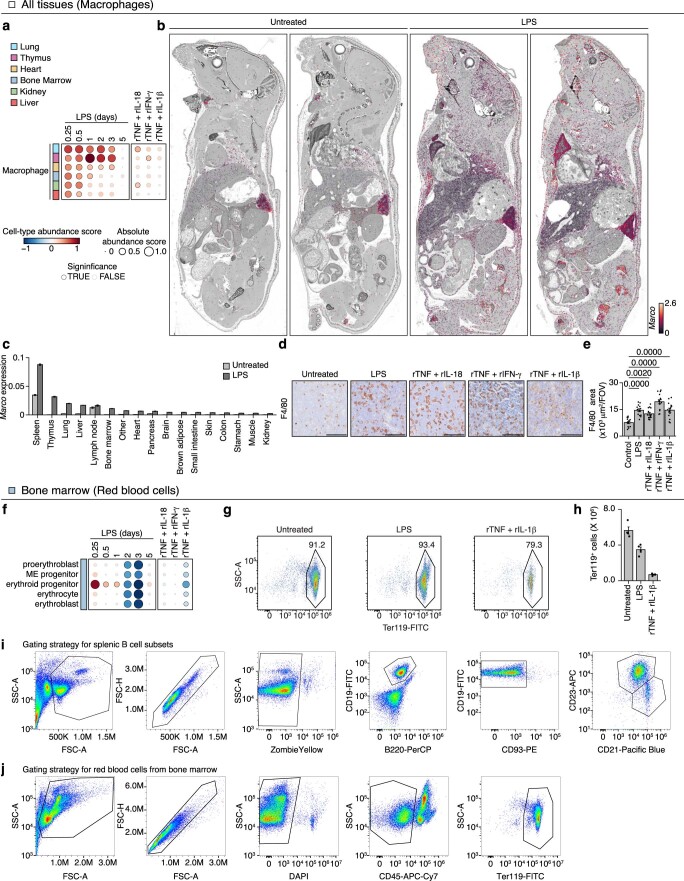
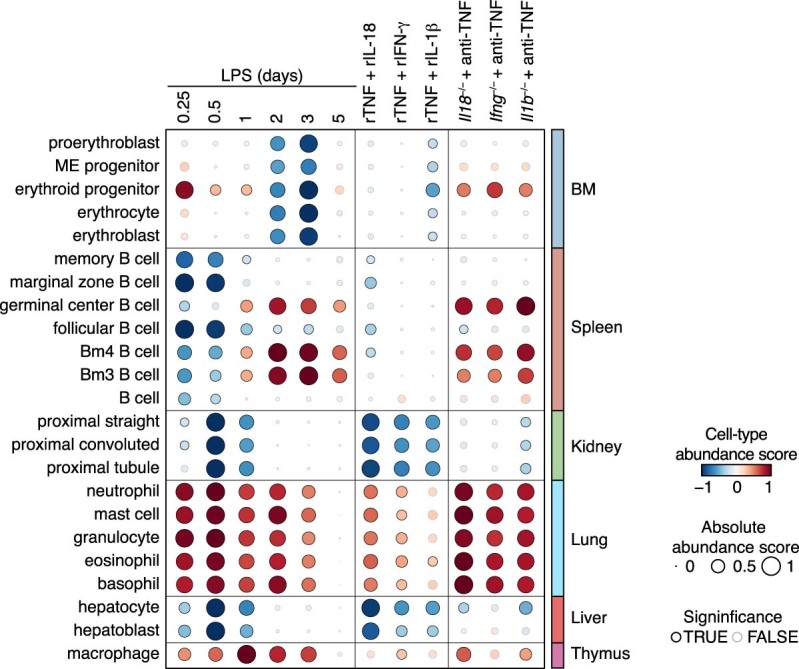
Sepsis is a systemic response to infection with life-threatening consequences. Our understanding of the molecular and cellular impact of sepsis across organs remains rudimentary. Here, we characterize the pathogenesis of sepsis by measuring dynamic changes in gene expression across organs. To pinpoint molecules controlling organ states in sepsis, we compare the effects of sepsis on organ gene expression to those of 6 singles and 15 pairs of recombinant cytokines. Strikingly, we find that the pairwise effects of tumor necrosis factor plus interleukin (IL)-18, interferon-gamma or IL-1β suffice to mirror the impact of sepsis across tissues. Mechanistically, we map the cellular effects of sepsis and cytokines by computing changes in the abundance of 195 cell types across 9 organs, which we validate by whole-mouse spatial profiling. Our work decodes the cytokine cacophony in sepsis into a pairwise cytokine message capturing the gene, cell and tissue responses of the host to the disease.
© 2024. The Author(s).
Conflict of interest statement
A.P. is the founder and CEO of Combinatics. All other authors declare no competing interests.
Figures





Update of
-
Organism-Wide Analysis of Sepsis Reveals Mechanisms of Systemic Inflammation.bioRxiv [Preprint]. 2023 Feb 2:2023.01.30.526342. doi: 10.1101/2023.01.30.526342. bioRxiv. 2023. Update in: Nat Immunol. 2024 Feb;25(2):226-239. doi: 10.1038/s41590-023-01722-8. PMID: 36778287 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
