

A fast, scalable and versatile tool for analysis of single-cell omics data
- PMID: 38191932
- PMCID: PMC10864184
- DOI: 10.1038/s41592-023-02139-9
A fast, scalable and versatile tool for analysis of single-cell omics data
Abstract
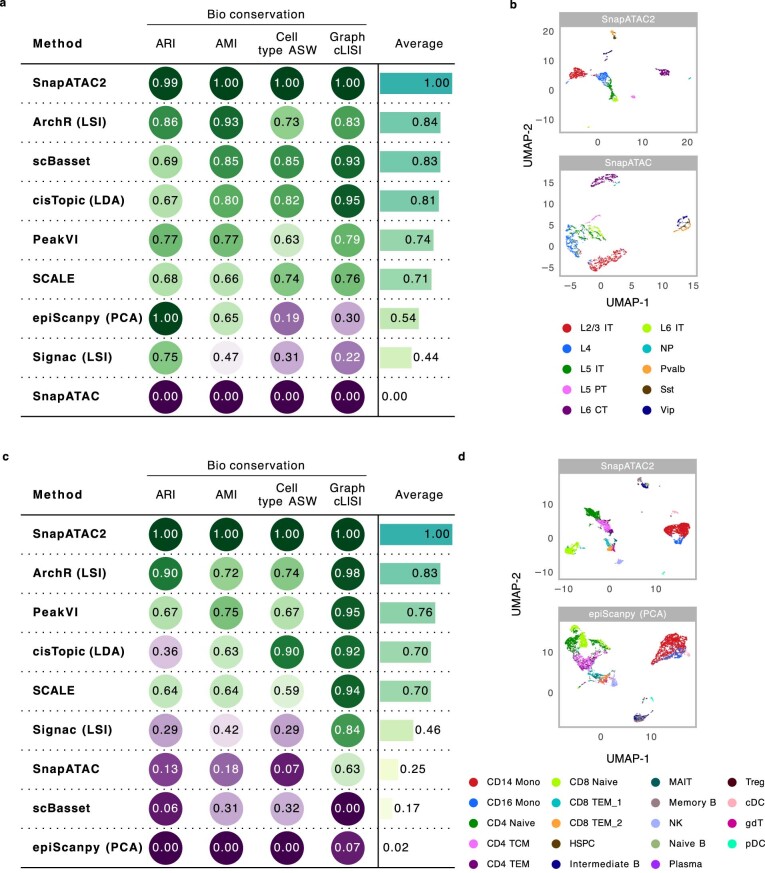
Single-cell omics technologies have revolutionized the study of gene regulation in complex tissues. A major computational challenge in analyzing these datasets is to project the large-scale and high-dimensional data into low-dimensional space while retaining the relative relationships between cells. This low dimension embedding is necessary to decompose cellular heterogeneity and reconstruct cell-type-specific gene regulatory programs. Traditional dimensionality reduction techniques, however, face challenges in computational efficiency and in comprehensively addressing cellular diversity across varied molecular modalities. Here we introduce a nonlinear dimensionality reduction algorithm, embodied in the Python package SnapATAC2, which not only achieves a more precise capture of single-cell omics data heterogeneities but also ensures efficient runtime and memory usage, scaling linearly with the number of cells. Our algorithm demonstrates exceptional performance, scalability and versatility across diverse single-cell omics datasets, including single-cell assay for transposase-accessible chromatin using sequencing, single-cell RNA sequencing, single-cell Hi-C and single-cell multi-omics datasets, underscoring its utility in advancing single-cell analysis.
© 2024. The Author(s).
Conflict of interest statement
B.R. is a cofounder of Epigenome Technologies, and a cofounder and consultant of Arima Genomics. The remaining authors declare no competing interests.
Figures

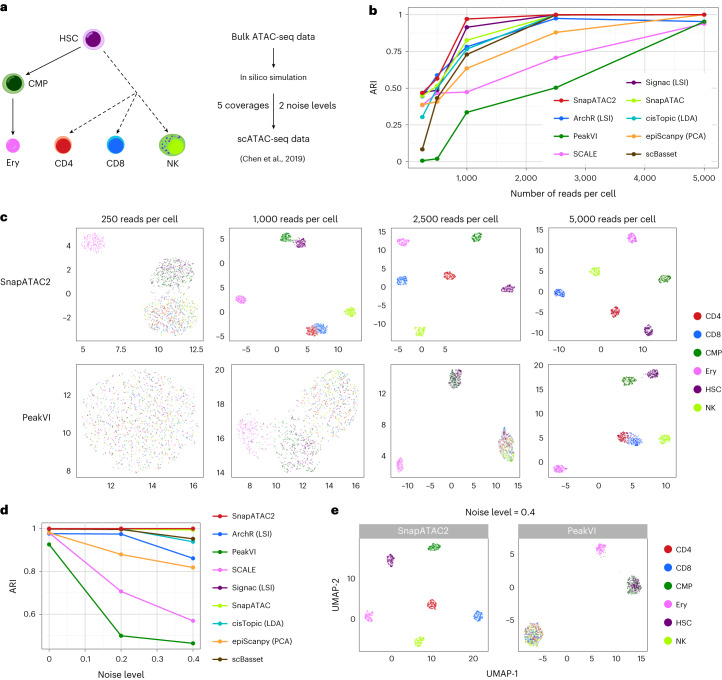
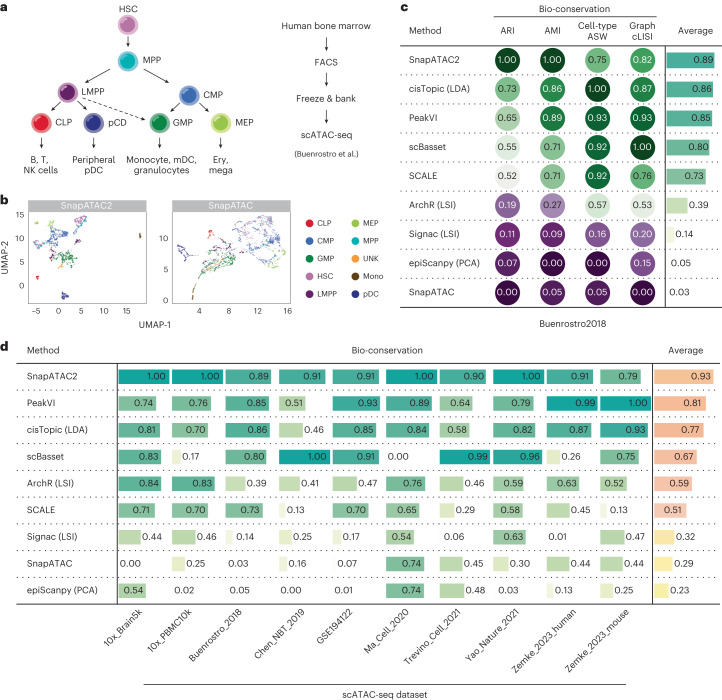
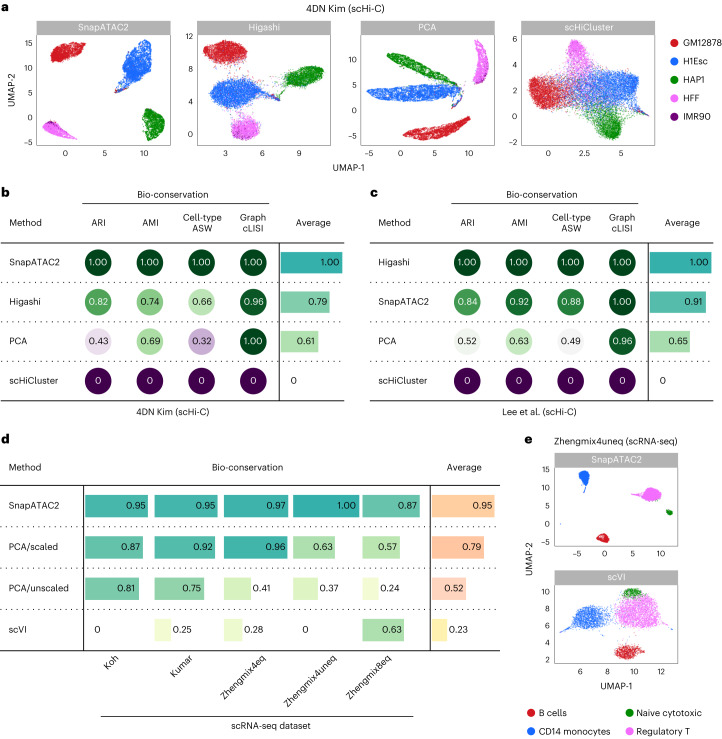
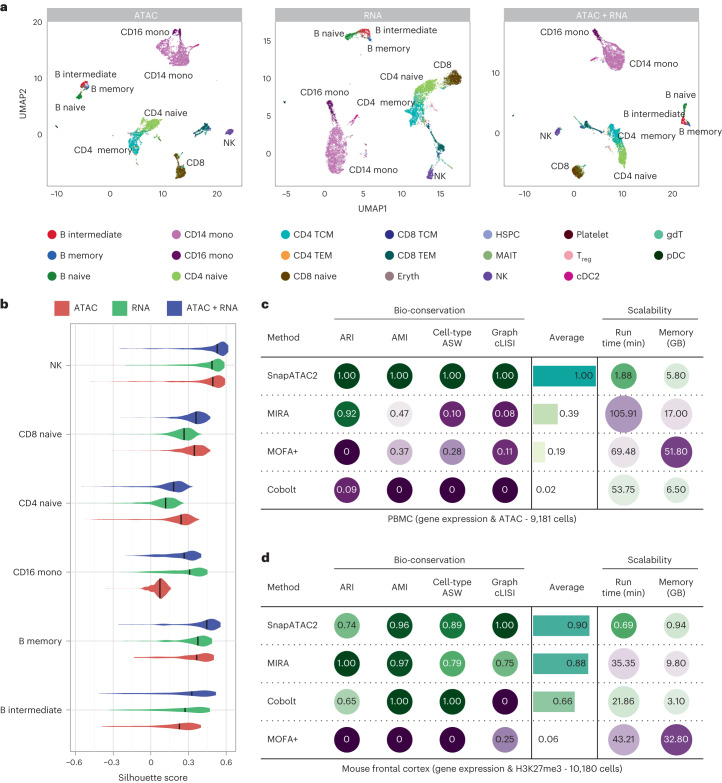









Update of
-
SnapATAC2: a fast, scalable and versatile tool for analysis of single-cell omics data.bioRxiv [Preprint]. 2023 Sep 15:2023.09.11.557221. doi: 10.1101/2023.09.11.557221. bioRxiv. 2023. Update in: Nat Methods. 2024 Feb;21(2):217-227. doi: 10.1038/s41592-023-02139-9. PMID: 37745443 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
