

Native Capillary Electrophoresis-Mass Spectrometry of Near 1 MDa Non-Covalent GroEL/GroES/Substrate Protein Complexes
- PMID: 38191978
- PMCID: PMC10953559
- DOI: 10.1002/advs.202306824
Native Capillary Electrophoresis-Mass Spectrometry of Near 1 MDa Non-Covalent GroEL/GroES/Substrate Protein Complexes
Abstract
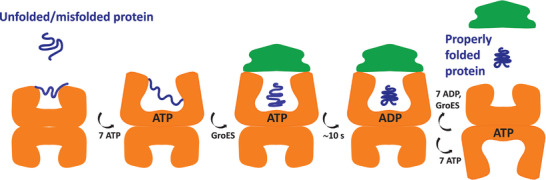
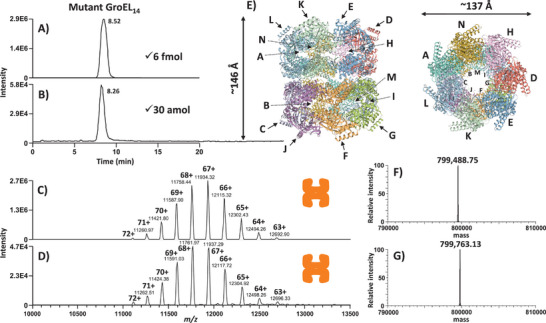
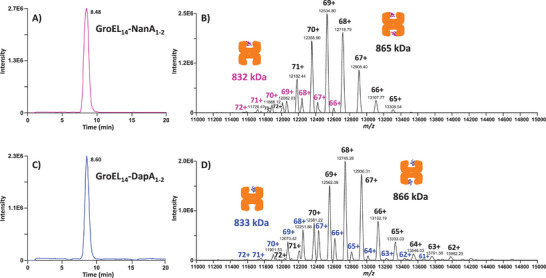
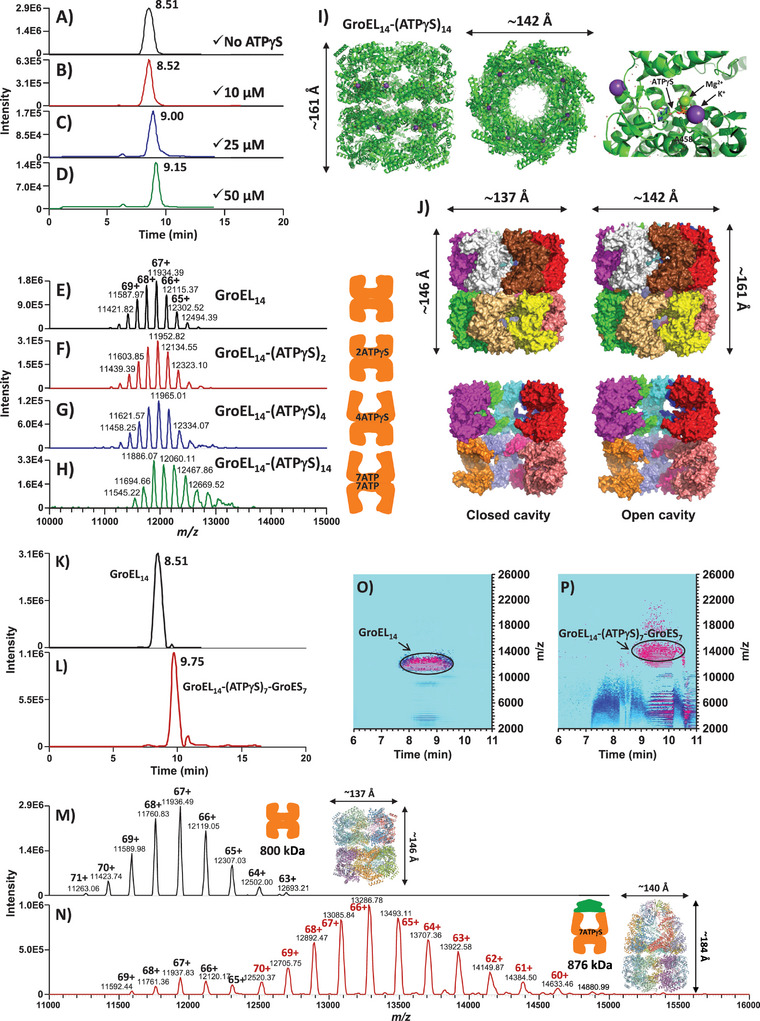
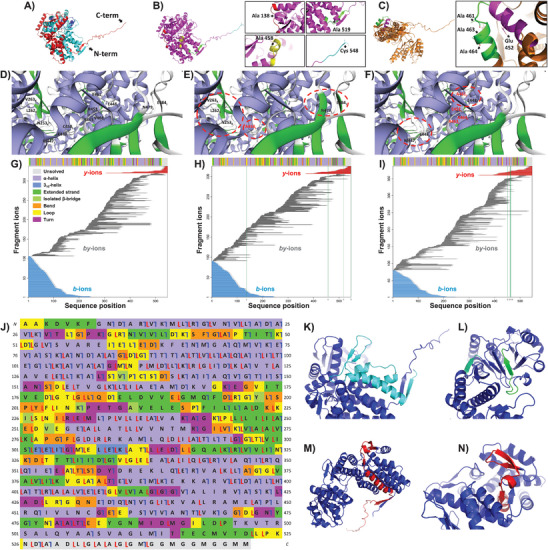
Protein complexes are essential for proteins' folding and biological function. Currently, native analysis of large multimeric protein complexes remains challenging. Structural biology techniques are time-consuming and often cannot monitor the proteins' dynamics in solution. Here, a capillary electrophoresis-mass spectrometry (CE-MS) method is reported to characterize, under near-physiological conditions, the conformational rearrangements of ∽1 MDa GroEL upon complexation with binding partners involved in a protein folding cycle. The developed CE-MS method is fast (30 min per run), highly sensitive (low-amol level), and requires ∽10 000-fold fewer samples compared to biochemical/biophysical techniques. The method successfully separates GroEL14 (∽800 kDa), GroEL7 (∽400 kDa), GroES7 (∽73 kDa), and NanA4 (∽130 kDa) oligomers. The non-covalent binding of natural substrate proteins with GroEL14 can be detected and quantified. The technique allows monitoring of GroEL14 conformational changes upon complexation with (ATPγS)4-14 and GroES7 (∽876 kDa). Native CE-pseudo-MS3 analyses of wild-type (WT) GroEL and two GroEL mutants result in up to 60% sequence coverage and highlight subtle structural differences between WT and mutated GroEL. The presented results demonstrate the superior CE-MS performance for multimeric complexes' characterization versus direct infusion ESI-MS. This study shows the CE-MS potential to provide information on binding stoichiometry and kinetics for various protein complexes.
Keywords: ATP-induced conformational rearrangement; GroEL; GroES; chaperones; native capillary electrophoresis-mass spectrometry.
© 2024 The Authors. Advanced Science published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Temperature Regulates Stability, Ligand Binding (Mg2+ and ATP), and Stoichiometry of GroEL-GroES Complexes.J Am Chem Soc. 2022 Feb 16;144(6):2667-2678. doi: 10.1021/jacs.1c11341. Epub 2022 Feb 2. J Am Chem Soc. 2022. PMID: 35107280 Free PMC article.
-
Conditions for nucleotide-dependent GroES-GroEL interactions. GroEL14(groES7)2 is favored by an asymmetric distribution of nucleotides.J Biol Chem. 1997 Oct 24;272(43):26999-7004. doi: 10.1074/jbc.272.43.26999. J Biol Chem. 1997. PMID: 9341138
-
Symmetric GroEL-GroES complexes can contain substrate simultaneously in both GroEL rings.FEBS Lett. 1997 Mar 24;405(2):195-9. doi: 10.1016/s0014-5793(97)00186-5. FEBS Lett. 1997. PMID: 9089290
-
Iterative annealing mechanism explains the functions of the GroEL and RNA chaperones.Protein Sci. 2020 Feb;29(2):360-377. doi: 10.1002/pro.3795. Epub 2019 Dec 23. Protein Sci. 2020. PMID: 31800116 Free PMC article. Review.
-
Protein folding assisted by the GroEL/GroES chaperonin system.Biochemistry (Mosc). 1998 Apr;63(4):374-81. Biochemistry (Mosc). 1998. PMID: 9556520 Review.
Cited by
-
Capillary Electrophoresis-Mass Spectrometry for Top-Down Proteomics.Annu Rev Anal Chem (Palo Alto Calif). 2025 May;18(1):125-147. doi: 10.1146/annurev-anchem-071124-092242. Epub 2025 Jan 23. Annu Rev Anal Chem (Palo Alto Calif). 2025. PMID: 39847747 Review.
-
Does Native Capillary Zone Electrophoresis-Mass Spectrometry Maintain the Structural Topology of Protein Complexes?Anal Chem. 2025 Apr 15;97(14):7616-7621. doi: 10.1021/acs.analchem.4c06949. Epub 2025 Apr 1. Anal Chem. 2025. PMID: 40167190 Free PMC article.
-
Top-Down Proteomic Analysis of Limited Samples Using Porous Layer Open Tubular Columns and High-Field Asymmetric Ion Mobility Spectrometry Coupled to Mass Spectrometry.Anal Chem. 2025 Jul 15;97(27):14189-14199. doi: 10.1021/acs.analchem.4c06727. Epub 2025 Jun 30. Anal Chem. 2025. PMID: 40586374 Free PMC article.
-
Native Proteomics by Capillary Zone Electrophoresis-Mass Spectrometry.Angew Chem Int Ed Engl. 2024 Nov 25;63(48):e202408370. doi: 10.1002/anie.202408370. Epub 2024 Oct 24. Angew Chem Int Ed Engl. 2024. PMID: 39196601
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials