

Protective effect of Tat fused HPCA protein on neuronal cell death caused by ischemic injury
- PMID: 38192804
- PMCID: PMC10772100
- DOI: 10.1016/j.heliyon.2023.e23488
Protective effect of Tat fused HPCA protein on neuronal cell death caused by ischemic injury
Abstract
Background: Bain ischemia is a disease that occurs for various reasons, induces reactive oxygen species (ROS), and causes fatal damage to the nervous system. Protective effect of HPCA on ischemic injury has not been extensively studied despite its significance in regulating calcium homeostasis and promoting neuronal survival in CA1 region of the brain.
Objective: We investigate the role of HPCA in ischemic injury using a cell-permeable Tat peptide fused HPCA protein (Tat-HPCA).
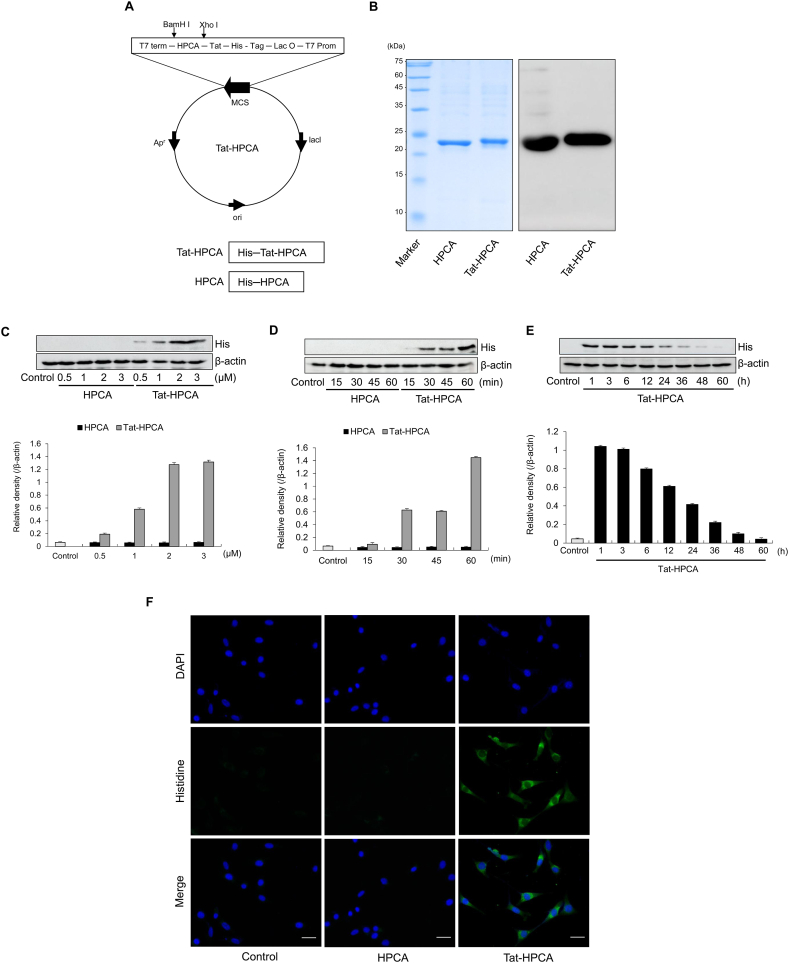
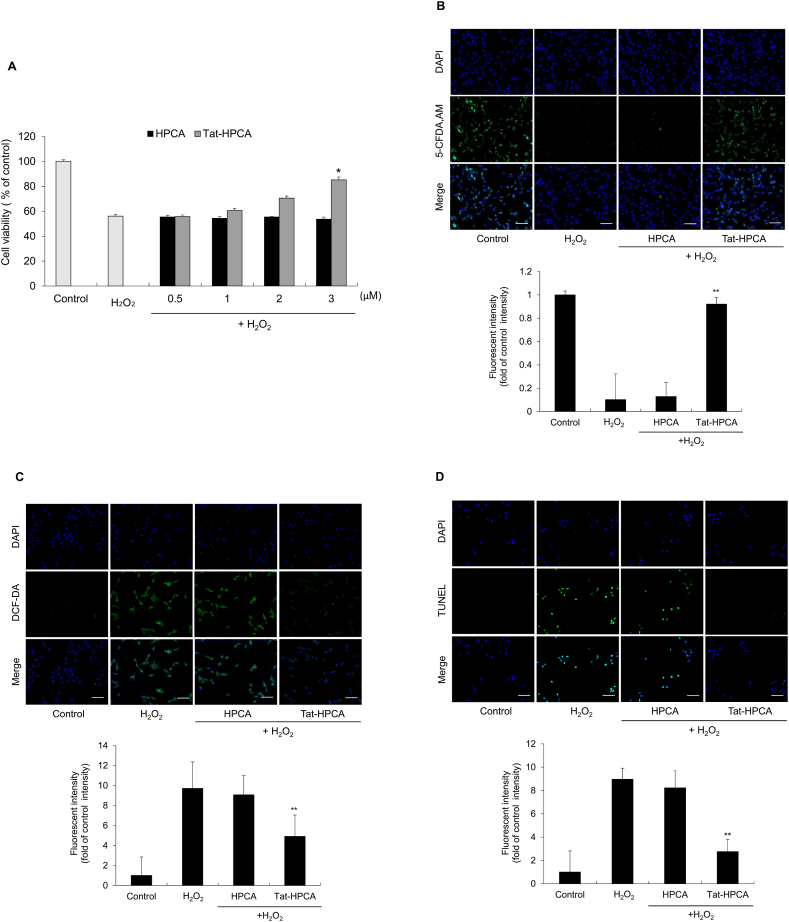
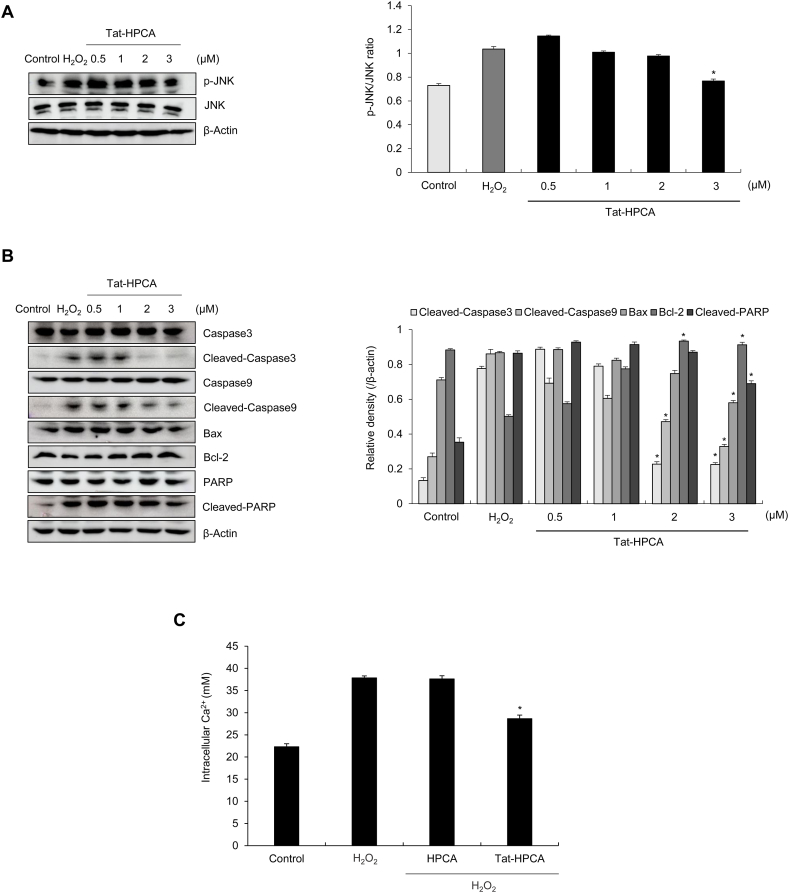
Methods: Western blot analysis determined the penetration of Tat-HPCA into HT-22 cells and apoptotic signaling pathways. 5-CFDA, AM, DCF-DA, and TUNEL staining confirmed intracellular ROS production and DNA damage. The intracellular Ca2+ was measured in primary cultured neurons treated with H2O2. Protective effects were examined using immunohistochemistry and cognitive function tests by passive avoidance test and 8-arm radial maze test.
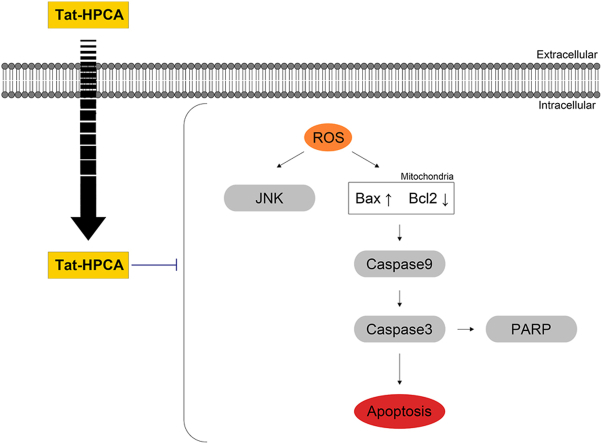
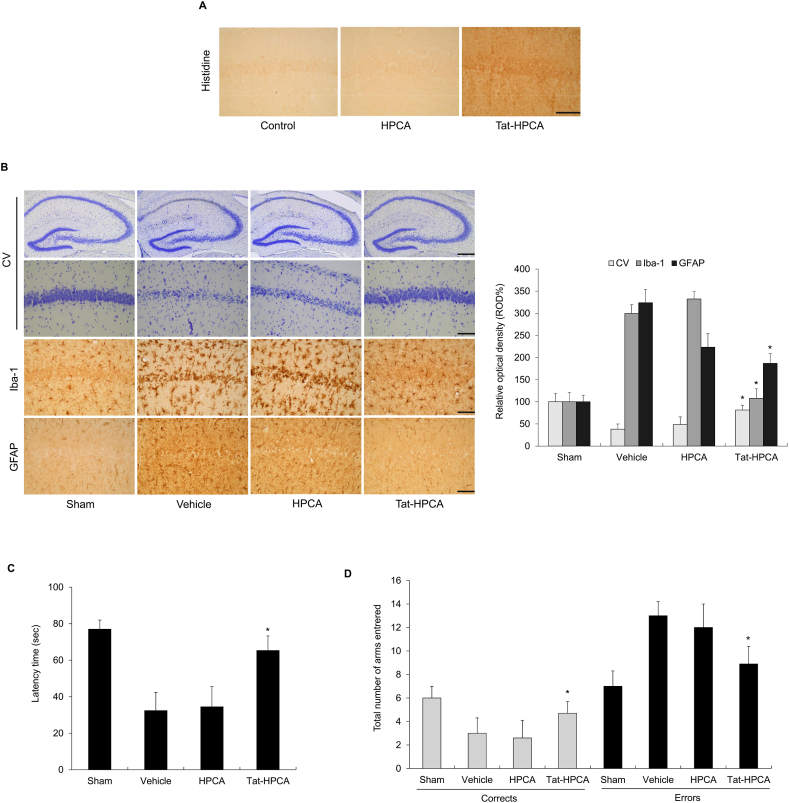
Results: Tat-HPCA effectively penetrated into HT-22 cells and inhibited H2O2-induced apoptosis, oxidative stress, and DNA fragmentation. It also effectively inhibited phosphorylation of JNK and regulated the activation of Caspase, Bax, Bcl-2, and PARP, leading to inhibition of apoptosis. Moreover, Ca2+ concentration decreased in cells treated with Tat-HPCA in primary cultured neurons. In an animal model of ischemia, Tat-HPCA effectively penetrated the hippocampus, inhibited cell death, and regulated activities of astrocytes and microglia. Additionally, Cognitive function tests show that Tat-HPCA improves neurobehavioral outcomes after cerebral ischemic injury.
Conclusion: These results suggest that Tat-HPCA might have potential as a therapeutic agent for treating oxidative stress-related diseases induced by ischemic injury, including ischemia.
Keywords: Brain ischemia; Oxidative stress; Protein therapy; Protein transduction domain; Tat-HPCA.
© 2023 The Authors.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Tat-NOL3 protects against hippocampal neuronal cell death induced by oxidative stress through the regulation of apoptotic pathways.Int J Mol Med. 2016 Jul;38(1):225-35. doi: 10.3892/ijmm.2016.2596. Epub 2016 May 19. Int J Mol Med. 2016. PMID: 27221790
-
Tat-antioxidant 1 protects against stress-induced hippocampal HT-22 cells death and attenuate ischaemic insult in animal model.J Cell Mol Med. 2015 Jun;19(6):1333-45. doi: 10.1111/jcmm.12513. Epub 2015 Mar 17. J Cell Mol Med. 2015. PMID: 25781353 Free PMC article.
-
Tat-CIAPIN1 inhibits hippocampal neuronal cell damage through the MAPK and apoptotic signaling pathways.Free Radic Biol Med. 2019 May 1;135:68-78. doi: 10.1016/j.freeradbiomed.2019.02.028. Epub 2019 Feb 25. Free Radic Biol Med. 2019. PMID: 30818058
-
Protective Role of Transduced Tat-Thioredoxin1 (Trx1) against Oxidative Stress-Induced Neuronal Cell Death via ASK1-MAPK Signal Pathway.Biomol Ther (Seoul). 2021 May 1;29(3):321-330. doi: 10.4062/biomolther.2020.154. Biomol Ther (Seoul). 2021. PMID: 33436533 Free PMC article.
-
Protective effects of cell permeable Tat-PIM2 protein on oxidative stress induced dopaminergic neuronal cell death.Heliyon. 2023 Apr 29;9(5):e15945. doi: 10.1016/j.heliyon.2023.e15945. eCollection 2023 May. Heliyon. 2023. PMID: 37223703 Free PMC article.
References
-
- Misra M.K., Sarwat M., Bhakuni P., Tuteja R., Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2009;15:RA209–RA219. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
