

Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy
- PMID: 38195537
- PMCID: PMC10775503
- DOI: 10.1186/s12943-023-01925-5
Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy
Erratum in
-
Correction: Comprehensive review of CRISPR‑based gene editing: mechanisms, challenges, and applications in cancer therapy.Mol Cancer. 2024 Feb 27;23(1):43. doi: 10.1186/s12943-024-01961-9. Mol Cancer. 2024. PMID: 38413951 Free PMC article. No abstract available.
Abstract
The CRISPR system is a revolutionary genome editing tool that has the potential to revolutionize the field of cancer research and therapy. The ability to precisely target and edit specific genetic mutations that drive the growth and spread of tumors has opened up new possibilities for the development of more effective and personalized cancer treatments. In this review, we will discuss the different CRISPR-based strategies that have been proposed for cancer therapy, including inactivating genes that drive tumor growth, enhancing the immune response to cancer cells, repairing genetic mutations that cause cancer, and delivering cancer-killing molecules directly to tumor cells. We will also summarize the current state of preclinical studies and clinical trials of CRISPR-based cancer therapy, highlighting the most promising results and the challenges that still need to be overcome. Safety and delivery are also important challenges for CRISPR-based cancer therapy to become a viable clinical option. We will discuss the challenges and limitations that need to be overcome, such as off-target effects, safety, and delivery to the tumor site. Finally, we will provide an overview of the current challenges and opportunities in the field of CRISPR-based cancer therapy and discuss future directions for research and development. The CRISPR system has the potential to change the landscape of cancer research, and this review aims to provide an overview of the current state of the field and the challenges that need to be overcome to realize this potential.
Keywords: CRISPR system; Cancer therapy; Cancer-killing molecules; Clinical trials; Delivery; Genetic mutations; Genome editing; Immune response; Off-target effects; Preclinical studies; Safety; Tumor growth.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
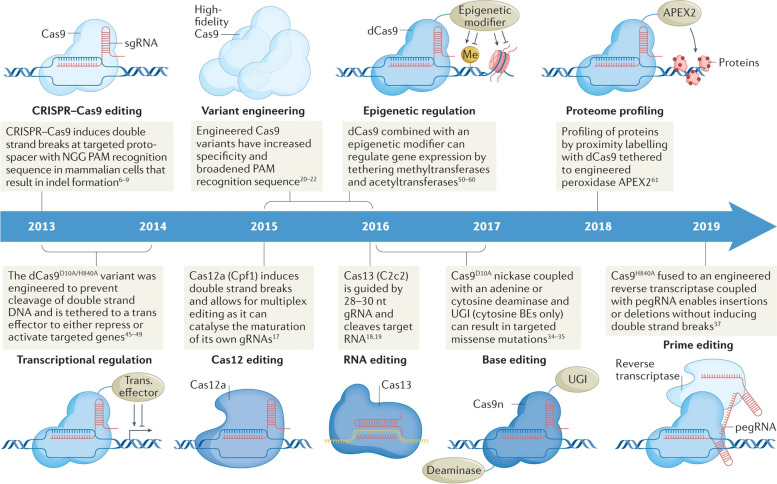
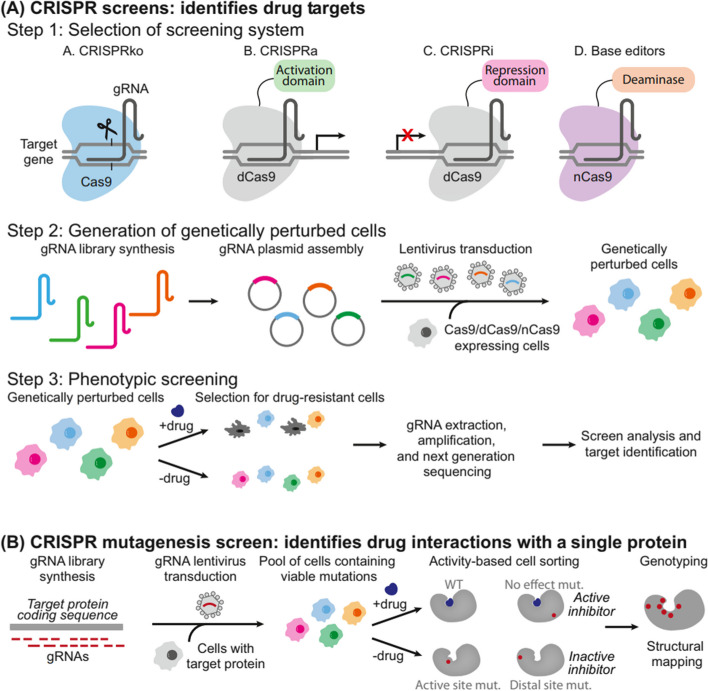
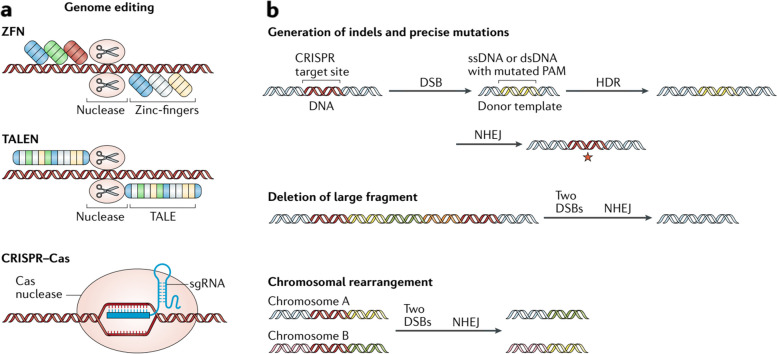

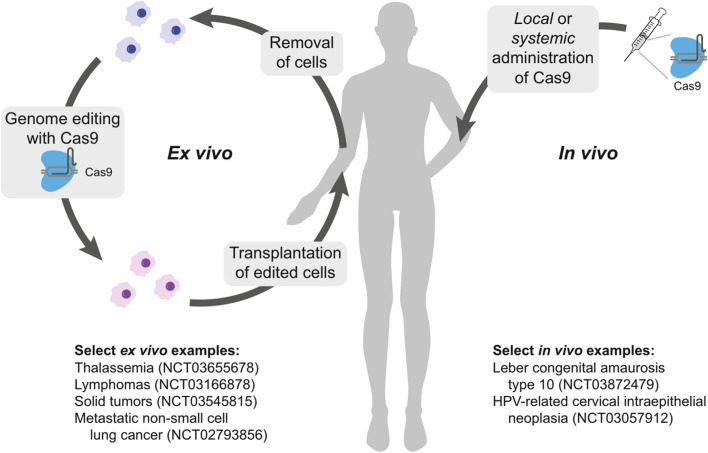
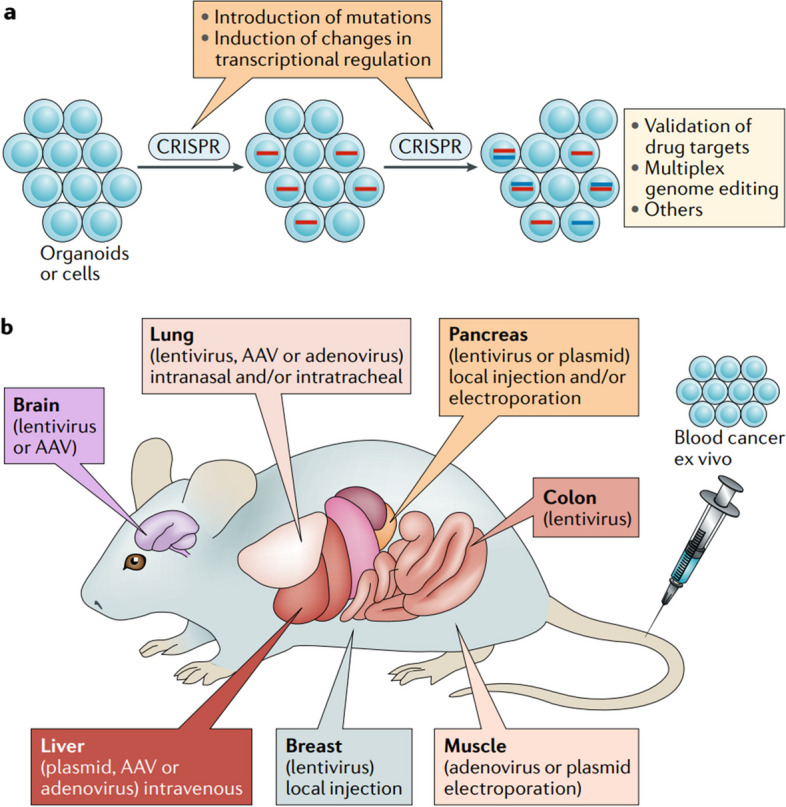
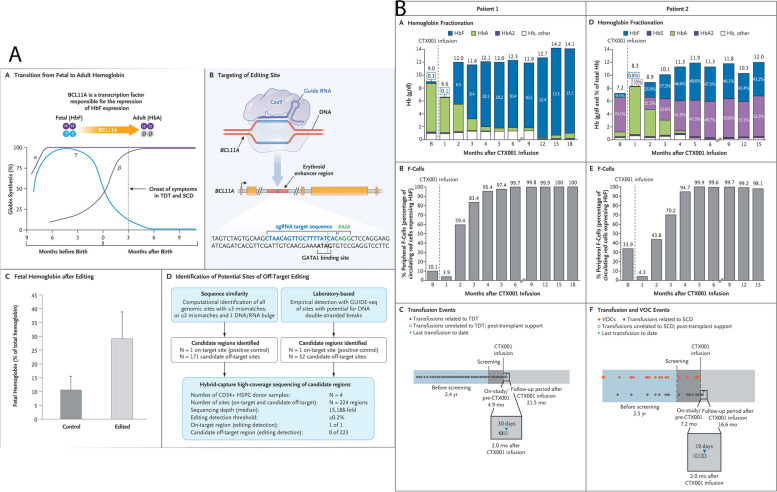
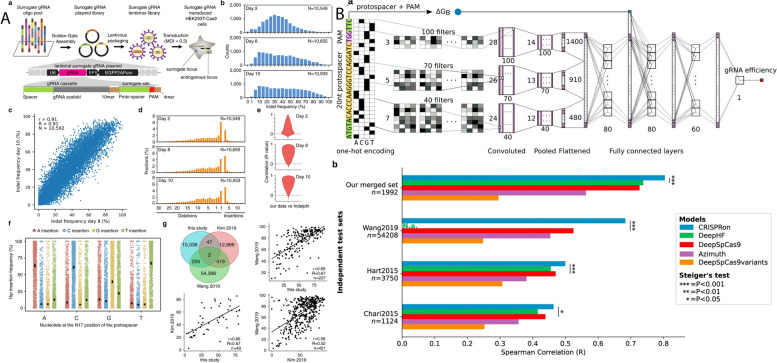

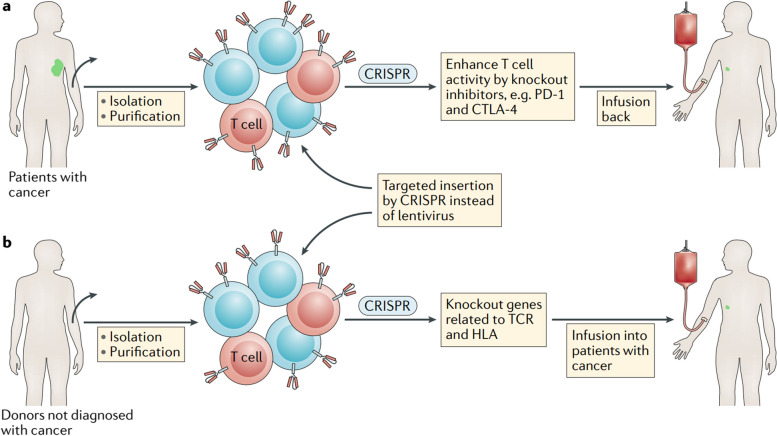

References
-
- De A, Biswas AR. Nanotechnology and Computational tool based study of CRISPR/Cas-9 research in Biomedical Engineering. J Nano Res Adv Mater Polym Sci. 2020;1:6–1.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
