

Single-molecule FRET probes allosteric effects on protein-translocating pore loops of a AAA+ machine
- PMID: 38196191
- PMCID: PMC10870172
- DOI: 10.1016/j.bpj.2024.01.002
Single-molecule FRET probes allosteric effects on protein-translocating pore loops of a AAA+ machine
Abstract
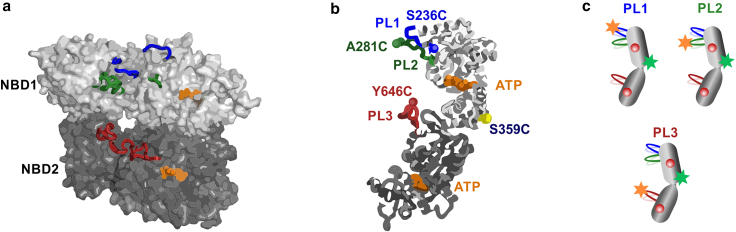
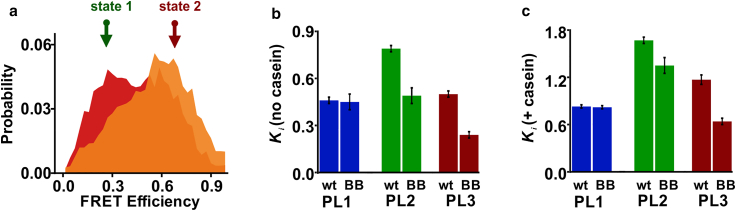
AAA+ proteins (ATPases associated with various cellular activities) comprise a family of powerful ring-shaped ATP-dependent translocases that carry out numerous vital substrate-remodeling functions. ClpB is a AAA+ protein disaggregation machine that forms a two-tiered hexameric ring, with flexible pore loops protruding into its center and binding to substrate proteins. It remains unknown whether these pore loops contribute only passively to substrate-protein threading or have a more active role. Recently, we have applied single-molecule FRET spectroscopy to directly measure the dynamics of substrate-binding pore loops in ClpB. We have reported that the three pore loops of ClpB (PL1-3) undergo large-scale fluctuations on the microsecond timescale that are likely to be mechanistically important for disaggregation. Here, using single-molecule FRET, we study the allosteric coupling between the pore loops and the two nucleotide-binding domains of ClpB (NBD1-2). By mutating the conserved Walker B motifs within the NBDs to abolish ATP hydrolysis, we demonstrate how the nucleotide state of each NBD tunes pore-loop dynamics. This effect is surprisingly long-ranged; in particular, PL2 and PL3 respond differentially to a Walker B mutation in either NBD1 or NBD2, as well as to mutations in both. We characterize the conformational dynamics of pore loops and the allosteric paths connecting NBDs to pore loops by molecular dynamics simulations and find that both principal motions and allosteric paths can be altered by changing the ATPase state of ClpB. Remarkably, PL3, which is highly conserved in AAA+ machines, is found to favor an upward conformation when only NBD1 undergoes ATP hydrolysis but a downward conformation when NBD2 is active. These results explicitly demonstrate a significant long-range allosteric effect of ATP hydrolysis sites on pore-loop dynamics. Pore loops are therefore established as active participants that undergo ATP-dependent conformational changes to translocate substrate proteins through the central pores of AAA+ machines.
Copyright © 2024 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures





References
-
- Crampton D.J., Mukherjee S., Richardson C.C. DNA-induced switch from independent to sequential dTTP hydrolysis in the bacteriophage T7 DNA helicase. Mol. Cell. 2006;21:165–174. - PubMed
-
- Enemark E.J., Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. - PubMed
-
- Neuwald A.F., Aravind L., et al. Koonin E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999;9:27–43. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
