

Improved detection and consistency of RNA-interacting proteomes using DIA SILAC
- PMID: 38197237
- PMCID: PMC10899761
- DOI: 10.1093/nar/gkad1249
Improved detection and consistency of RNA-interacting proteomes using DIA SILAC
Abstract
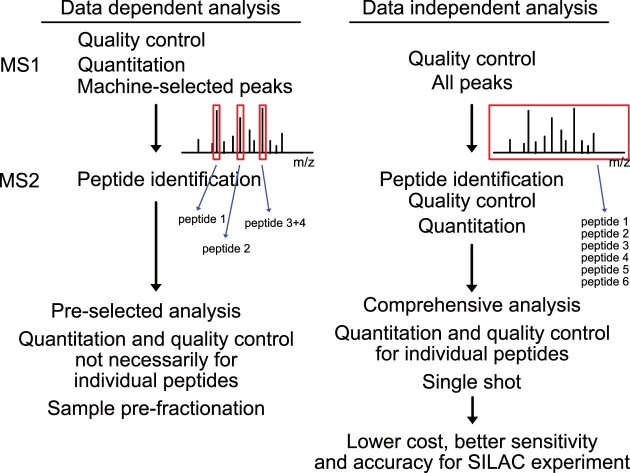
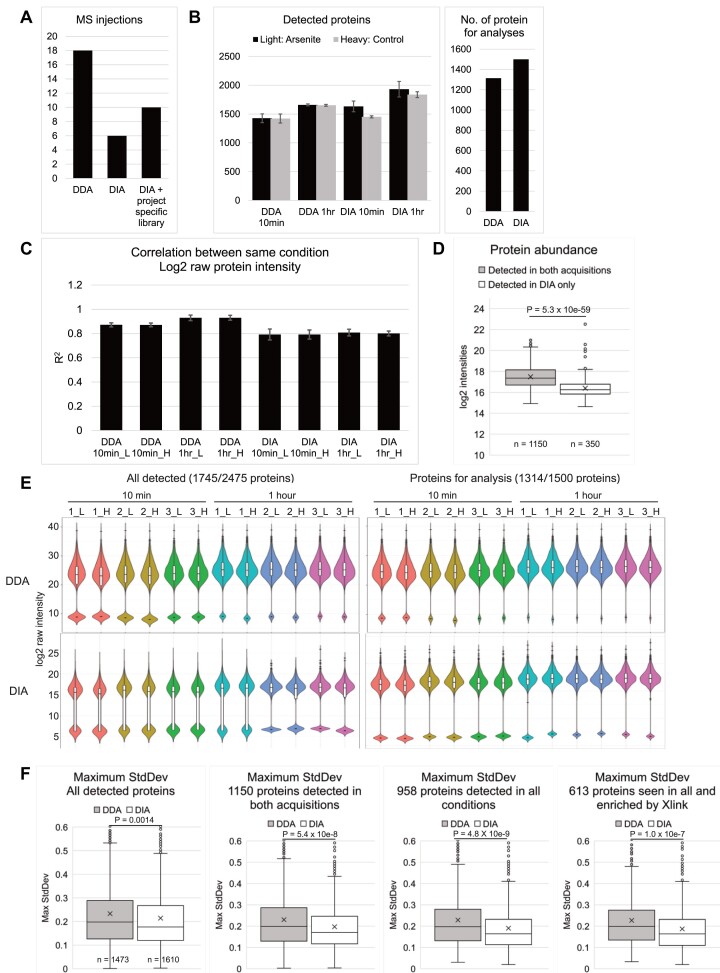
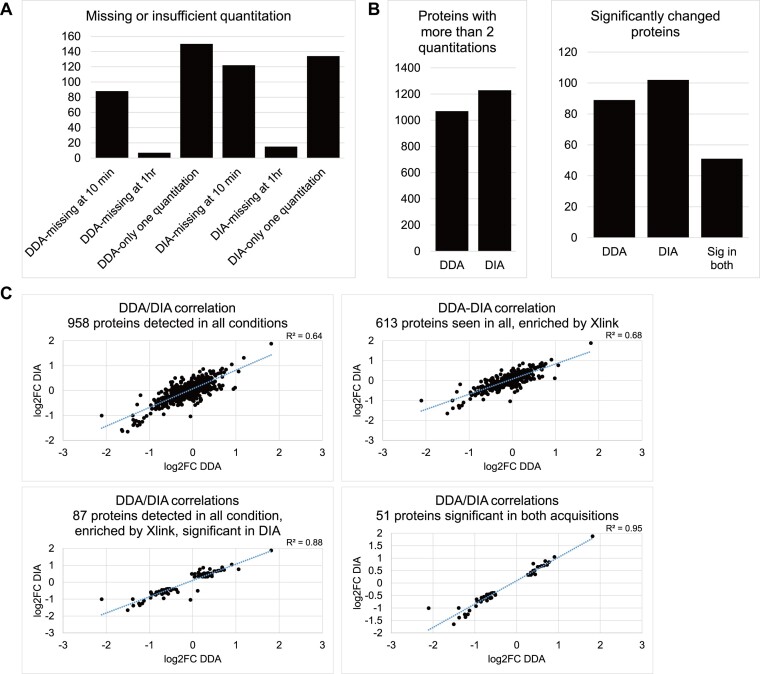
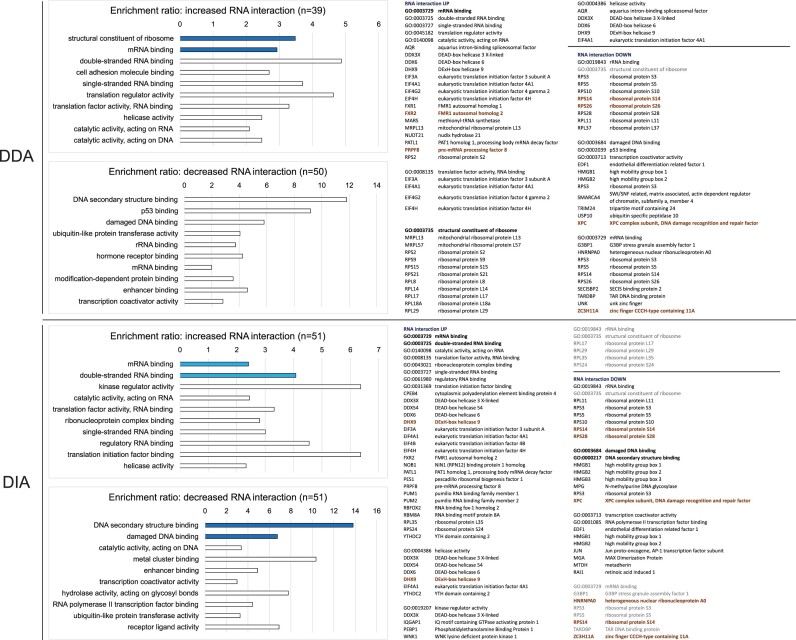
The RNA-interacting proteome is commonly characterized by UV-crosslinking followed by RNA purification, with protein recovery quantified using SILAC labeling followed by data-dependent acquisition (DDA) of proteomic data. However, the low efficiency of UV-crosslinking, combined with limited sensitivity of the DDA approach often restricts detection to relatively abundant proteins, necessitating multiple mass spec injections of fractionated peptides for each biological sample. Here we report an application of data-independent acquisition (DIA) with SILAC in a total RNA-associated protein purification (TRAPP) UV-crosslinking experiment. This gave 15% greater protein detection and lower inter-replicate variation relative to the same biological materials analyzed using DDA, while allowing single-shot analysis of the sample. As proof of concept, we determined the effects of arsenite treatment on the RNA-bound proteome of HEK293T cells. The DIA dataset yielded similar GO term enrichment for RNA-binding proteins involved in cellular stress responses to the DDA dataset while detecting extra proteins unseen by DDA. Overall, the DIA SILAC approach improved detection of proteins over conventional DDA SILAC for generating RNA-interactome datasets, at a lower cost due to reduced machine time. Analyses are described for TRAPP data, but the approach is suitable for proteomic analyses following essentially any RNA-binding protein enrichment technique.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures
Similar articles
-
Using SILAC to Develop Quantitative Data-Independent Acquisition (DIA) Proteomic Methods.Methods Mol Biol. 2023;2603:245-257. doi: 10.1007/978-1-0716-2863-8_20. Methods Mol Biol. 2023. PMID: 36370285
-
DIA-SIFT: A Precursor and Product Ion Filter for Accurate Stable Isotope Data-Independent Acquisition Proteomics.Anal Chem. 2018 Aug 7;90(15):8722-8726. doi: 10.1021/acs.analchem.8b01618. Epub 2018 Jul 19. Anal Chem. 2018. PMID: 29989796 Free PMC article.
-
Characterization of Cerebrospinal Fluid via Data-Independent Acquisition Mass Spectrometry.J Proteome Res. 2018 Oct 5;17(10):3418-3430. doi: 10.1021/acs.jproteome.8b00308. Epub 2018 Sep 12. J Proteome Res. 2018. PMID: 30207155
-
Improving Phosphoproteomics Profiling Using Data-Independent Mass Spectrometry.J Proteome Res. 2022 Aug 5;21(8):1789-1799. doi: 10.1021/acs.jproteome.2c00172. Epub 2022 Jul 25. J Proteome Res. 2022. PMID: 35877786 Review.
-
[Data-driven mass spectrometry methods (DDA) and data-independent methods (DIA) used in the analysis of biological material].Postepy Biochem. 2024 Jul 1;70(2):212-222. doi: 10.18388/pb.2021_535. Print 2024 Jul 1. Postepy Biochem. 2024. PMID: 39083467 Review. Polish.
Cited by
-
Data acquisition approaches for single cell proteomics.Proteomics. 2025 Jan;25(1-2):e2400022. doi: 10.1002/pmic.202400022. Epub 2024 Aug 1. Proteomics. 2025. PMID: 39088833 Free PMC article. Review.
References
-
- Queiroz R.M.L., Smith T., Villanueva E., Marti-Solano M., Monti M., Pizzinga M., Mirea D.-M., Ramakrishna M., Harvey R.F., Dezi V. et al. . Comprehensive identification of RNA–protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotech. 2019; 37:169–178. - PMC - PubMed
-
- Trendel J., Schwarzl T., Horos R., Prakash A., Bateman A., Hentze M.W., Krijgsveld J. The human RNA-binding proteome and its dynamics during translational arrest. Cell. 2019; 176:391–403. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
