

Quantification of biases in predictions of protein-protein binding affinity changes upon mutations
- PMID: 38197311
- PMCID: PMC10777193
- DOI: 10.1093/bib/bbad491
Quantification of biases in predictions of protein-protein binding affinity changes upon mutations
Abstract
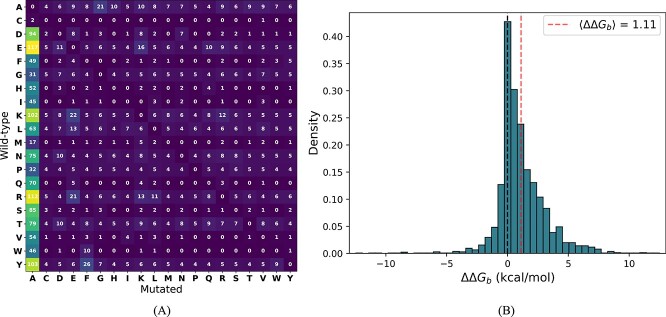
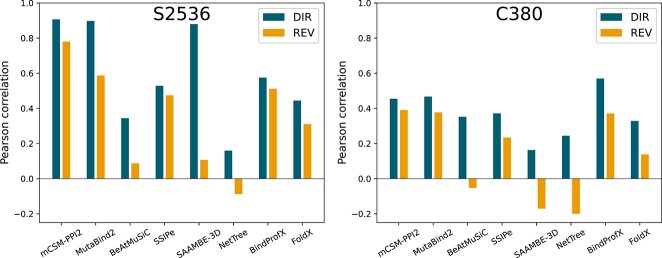
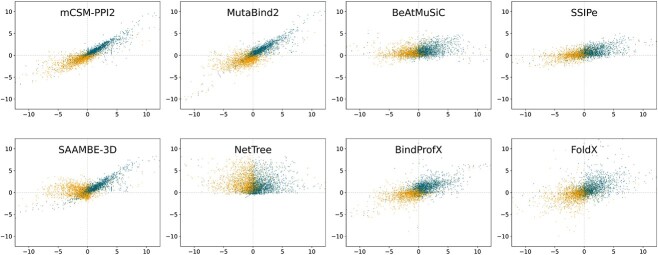
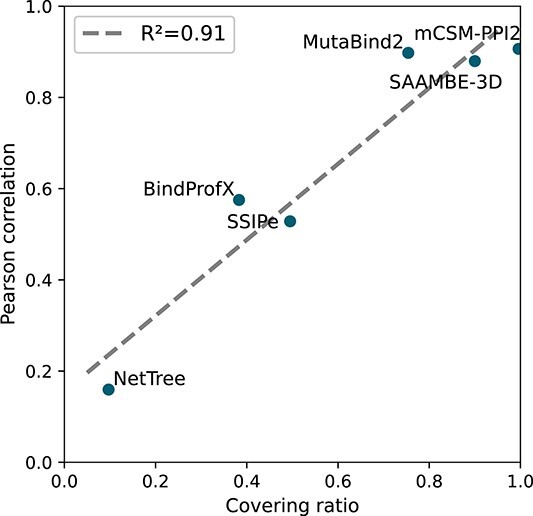
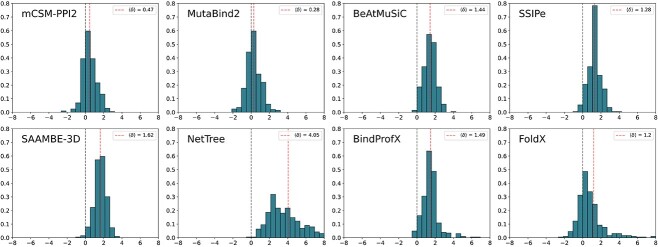
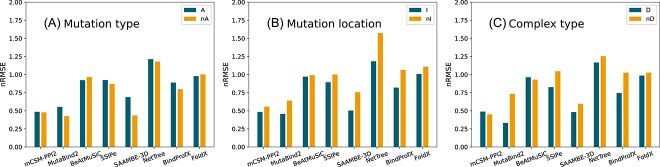
Understanding the impact of mutations on protein-protein binding affinity is a key objective for a wide range of biotechnological applications and for shedding light on disease-causing mutations, which are often located at protein-protein interfaces. Over the past decade, many computational methods using physics-based and/or machine learning approaches have been developed to predict how protein binding affinity changes upon mutations. They all claim to achieve astonishing accuracy on both training and test sets, with performances on standard benchmarks such as SKEMPI 2.0 that seem overly optimistic. Here we benchmarked eight well-known and well-used predictors and identified their biases and dataset dependencies, using not only SKEMPI 2.0 as a test set but also deep mutagenesis data on the severe acute respiratory syndrome coronavirus 2 spike protein in complex with the human angiotensin-converting enzyme 2. We showed that, even though most of the tested methods reach a significant degree of robustness and accuracy, they suffer from limited generalizability properties and struggle to predict unseen mutations. Interestingly, the generalizability problems are more severe for pure machine learning approaches, while physics-based methods are less affected by this issue. Moreover, undesirable prediction biases toward specific mutation properties, the most marked being toward destabilizing mutations, are also observed and should be carefully considered by method developers. We conclude from our analyses that there is room for improvement in the prediction models and suggest ways to check, assess and improve their generalizability and robustness.
Keywords: machine learning; prediction biases; protein complex structure; protein–protein binding affinity; protein–protein interactions; symmetry principle.
© The Author(s) 2024. Published by Oxford University Press.
Figures
Similar articles
-
Quantification of biases in predictions of protein stability changes upon mutations.Bioinformatics. 2018 Nov 1;34(21):3659-3665. doi: 10.1093/bioinformatics/bty348. Bioinformatics. 2018. PMID: 29718106
-
iSEE: Interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations.Proteins. 2019 Feb;87(2):110-119. doi: 10.1002/prot.25630. Epub 2018 Dec 3. Proteins. 2019. PMID: 30417935 Free PMC article.
-
D3AI-Spike: A deep learning platform for predicting binding affinity between SARS-CoV-2 spike receptor binding domain with multiple amino acid mutations and human angiotensin-converting enzyme 2.Comput Biol Med. 2022 Dec;151(Pt A):106212. doi: 10.1016/j.compbiomed.2022.106212. Epub 2022 Oct 25. Comput Biol Med. 2022. PMID: 36327885 Free PMC article.
-
Computational modeling of the effect of five mutations on the structure of the ACE2 receptor and their correlation with infectivity and virulence of some emerged variants of SARS-CoV-2 suggests mechanisms of binding affinity dysregulation.Chem Biol Interact. 2022 Dec 1;368:110244. doi: 10.1016/j.cbi.2022.110244. Epub 2022 Nov 3. Chem Biol Interact. 2022. PMID: 36336003 Free PMC article.
-
Artificial intelligence challenges for predicting the impact of mutations on protein stability.Curr Opin Struct Biol. 2022 Feb;72:161-168. doi: 10.1016/j.sbi.2021.11.001. Epub 2021 Dec 15. Curr Opin Struct Biol. 2022. PMID: 34922207 Review.
Cited by
-
AlphaFold2-Enabled Atomistic Modeling of Structure, Conformational Ensembles, and Binding Energetics of the SARS-CoV-2 Omicron BA.2.86 Spike Protein with ACE2 Host Receptor and Antibodies: Compensatory Functional Effects of Binding Hotspots in Modulating Mechanisms of Receptor Binding and Immune Escape.J Chem Inf Model. 2024 Mar 11;64(5):1657-1681. doi: 10.1021/acs.jcim.3c01857. Epub 2024 Feb 19. J Chem Inf Model. 2024. PMID: 38373700 Free PMC article.
-
Graph masked self-distillation learning for prediction of mutation impact on protein-protein interactions.Commun Biol. 2024 Oct 26;7(1):1400. doi: 10.1038/s42003-024-07066-9. Commun Biol. 2024. PMID: 39462102 Free PMC article.
-
Deciphering GB1's Single Mutational Landscape: Insights from MuMi Analysis.J Phys Chem B. 2024 Aug 22;128(33):7987-7996. doi: 10.1021/acs.jpcb.4c04916. Epub 2024 Aug 8. J Phys Chem B. 2024. PMID: 39115184 Free PMC article.
-
MPA-MutPred: a novel strategy for accurately predicting the binding affinity change upon mutation in membrane protein complexes.Brief Bioinform. 2024 Sep 23;25(6):bbae598. doi: 10.1093/bib/bbae598. Brief Bioinform. 2024. PMID: 39550225 Free PMC article.
-
Guidelines for releasing a variant effect predictor.ArXiv [Preprint]. 2024 Apr 16:arXiv:2404.10807v1. ArXiv. 2024. Update in: Genome Biol. 2025 Apr 15;26(1):97. doi: 10.1186/s13059-025-03572-z. PMID: 38699161 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
