

Noninvasive Detection of Neuroendocrine Prostate Cancer through Targeted Cell-free DNA Methylation
- PMID: 38197680
- PMCID: PMC10905672
- DOI: 10.1158/2159-8290.CD-23-0754
Noninvasive Detection of Neuroendocrine Prostate Cancer through Targeted Cell-free DNA Methylation
Abstract
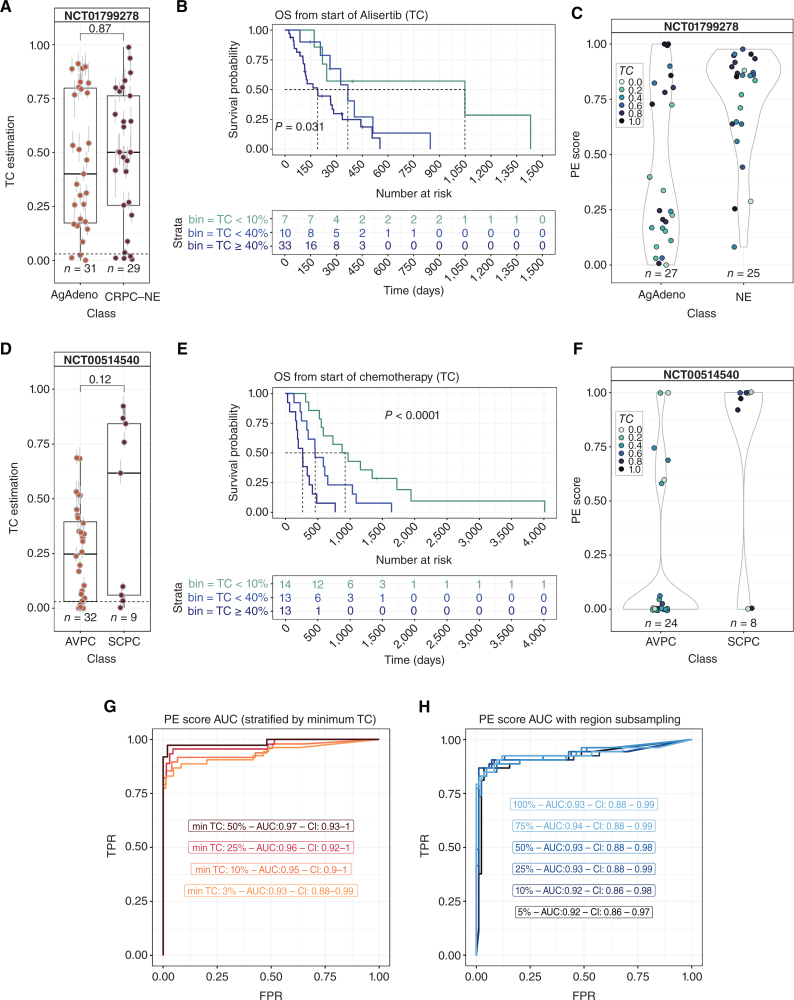
Castration-resistant prostate cancer (CRPC) is a heterogeneous disease associated with phenotypic subtypes that drive therapy response and outcome differences. Histologic transformation to castration-resistant neuroendocrine prostate cancer (CRPC-NE) is associated with distinct epigenetic alterations, including changes in DNA methylation. The current diagnosis of CRPC-NE is challenging and relies on metastatic biopsy. We developed a targeted DNA methylation assay to detect CRPC-NE using plasma cell-free DNA (cfDNA). The assay quantifies tumor content and provides a phenotype evidence score that captures diverse CRPC phenotypes, leveraging regions to inform transcriptional state. We tested the design in independent clinical cohorts (n = 222 plasma samples) and qualified it achieving an AUC > 0.93 for detecting pathology-confirmed CRPC-NE (n = 136). Methylation-defined cfDNA tumor content was associated with clinical outcomes in two prospective phase II clinical trials geared towards aggressive variant CRPC and CRPC-NE. These data support the application of targeted DNA methylation for CRPC-NE detection and patient stratification.
Significance: Neuroendocrine prostate cancer is an aggressive subtype of treatment-resistant prostate cancer. Early detection is important, but the diagnosis currently relies on metastatic biopsy. We describe the development and validation of a plasma cell-free DNA targeted methylation panel that can quantify tumor fraction and identify patients with neuroendocrine prostate cancer noninvasively. This article is featured in Selected Articles from This Issue, p. 384.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures





References
-
- Lorenzin F, Demichelis F. Evolution of the prostate cancer genome towards resistance. J Transl Genet Genom 2019;3:5.
-
- Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol 2018;15:271–86. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
