

CRIP1 involves the pathogenesis of multiple myeloma via dual-regulation of proteasome and autophagy
- PMID: 38199044
- PMCID: PMC10825369
- DOI: 10.1016/j.ebiom.2023.104961
CRIP1 involves the pathogenesis of multiple myeloma via dual-regulation of proteasome and autophagy
Abstract
Background: Multiple myeloma (MM) is an incurable hematological malignancy of the plasma cells. The maintenance of protein homeostasis is critical for MM cell survival. Elevated levels of paraproteins in MM cells are cleared by proteasomes or lysosomes, which are independent but inter-connected with each other. Proteasome inhibitors (PIs) work as a backbone agent and successfully improved the outcome of patients; however, the increasing activity of autophagy suppresses the sensitivity to PIs treatment.
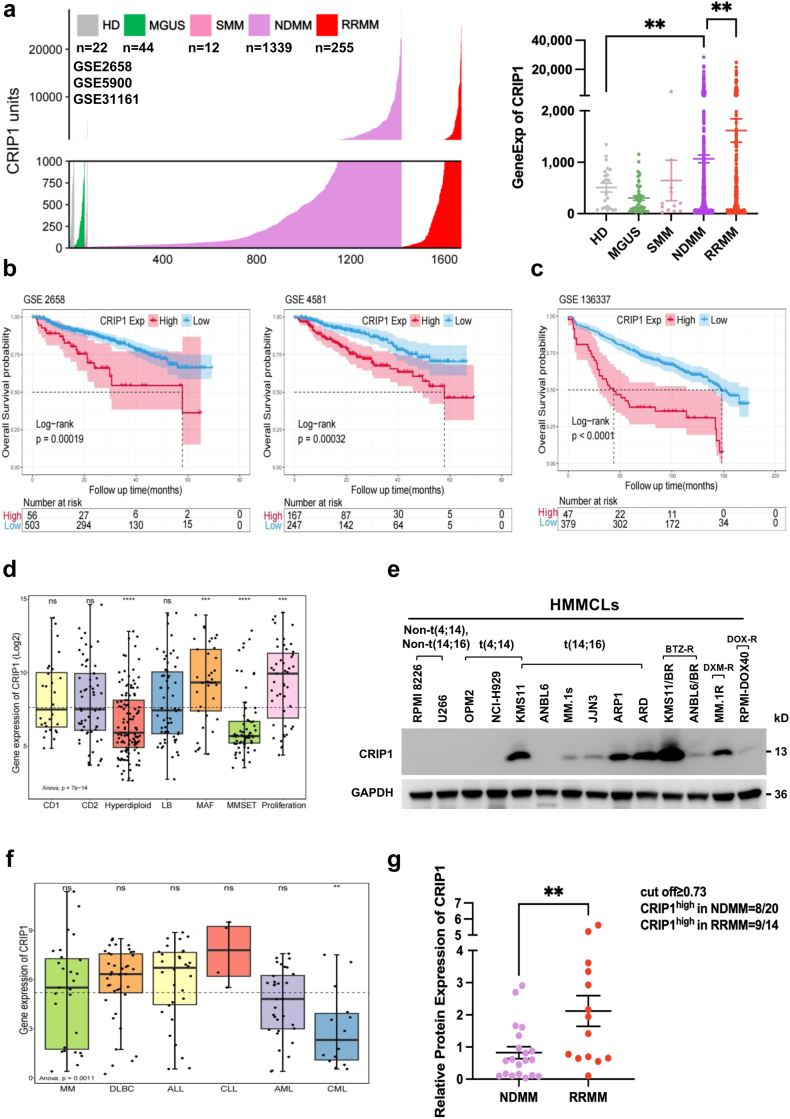
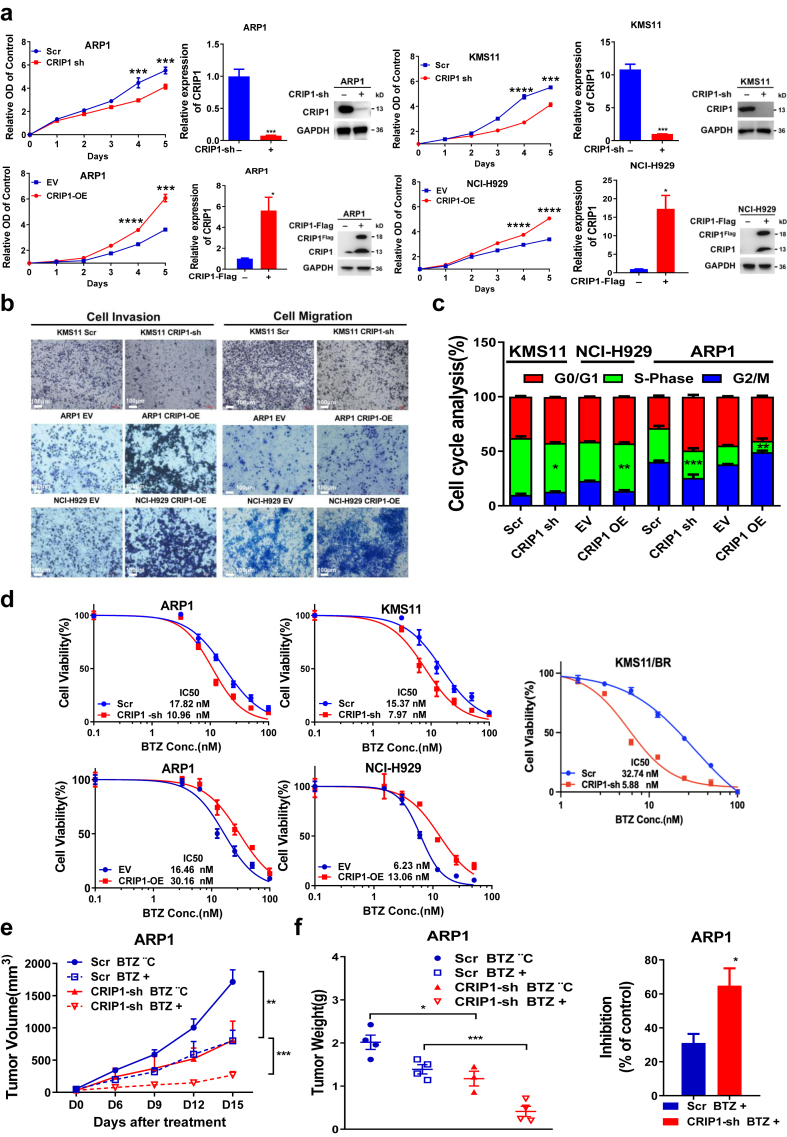
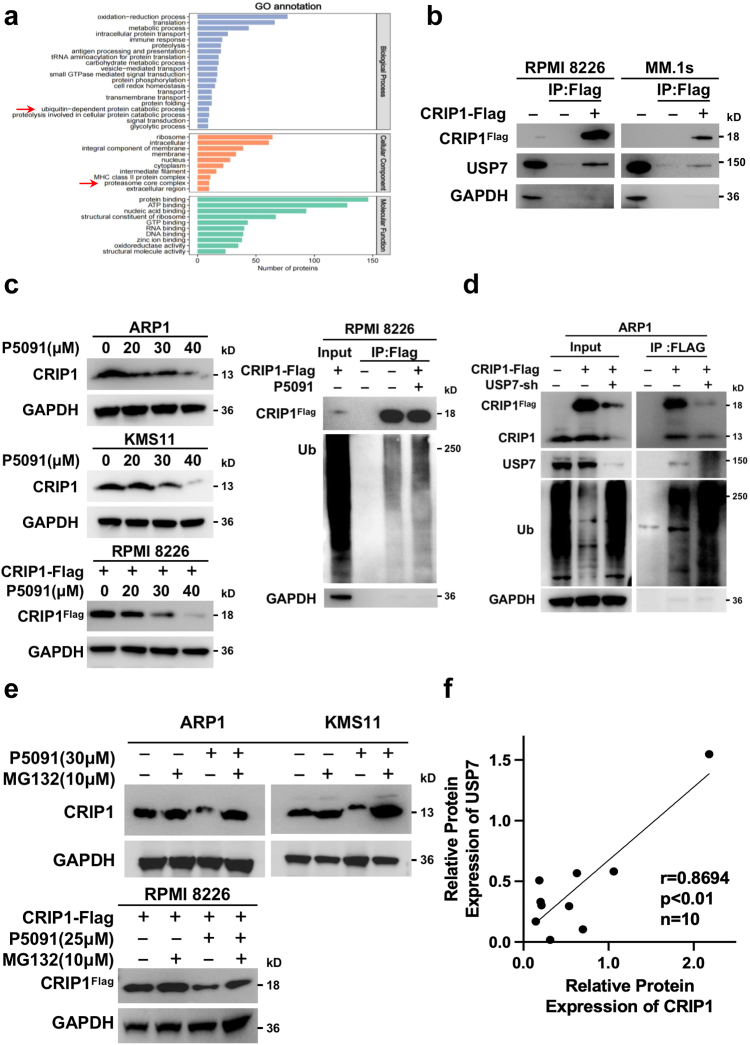
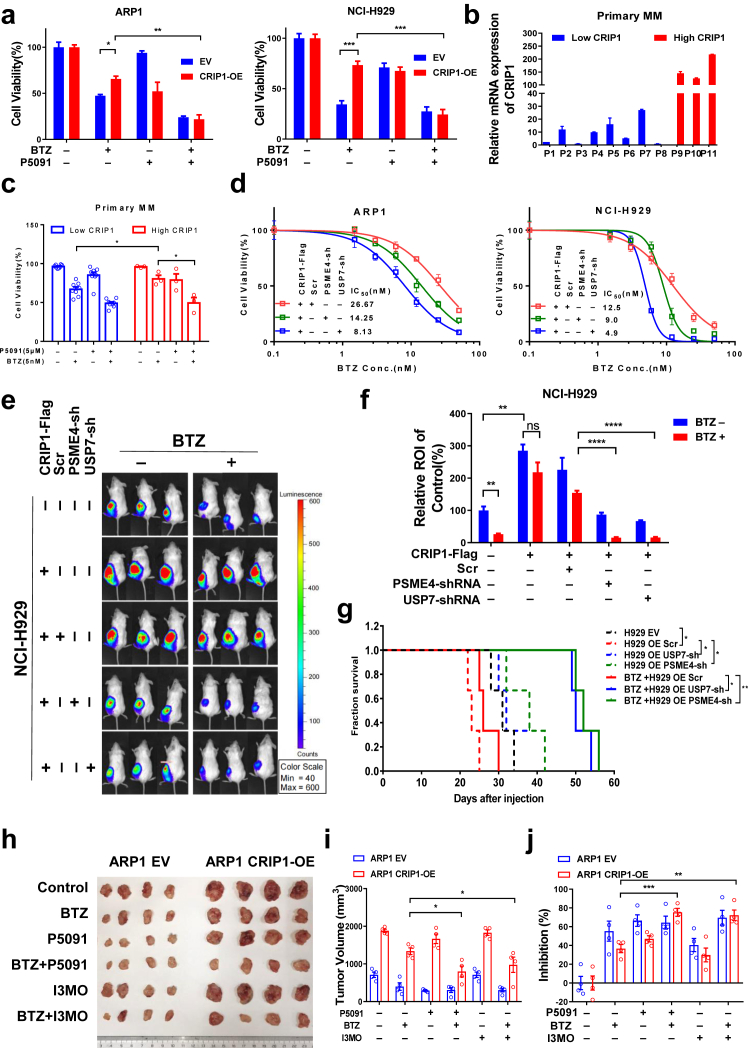
Methods: The transcription levels of CRIP1 were explored in plasma cells obtained from healthy donors, patients with newly diagnosed multiple myeloma (NDMM), and relapsed/refractory multiple myeloma (RRMM) using Gene expression omnibus datasets. Doxycycline-inducible CRIP1-shRNA and CRIP1 overexpressed MM cell lines were constructed to explore the role of CRIP1 in MM pathogenesis. Proliferation, invasion, migration, proteasome activity and autophagy were examined in MM cells with different CRIP1 levels. Co-immunoprecipitation (Co-IP) with Tandem affinity purification/Mass spectrum (TAP/MS) was performed to identify the binding proteins of CRIP1. The mouse xenograft model was used to determine the role of CRIP1 in the proliferation and drug-resistance of MM cells.
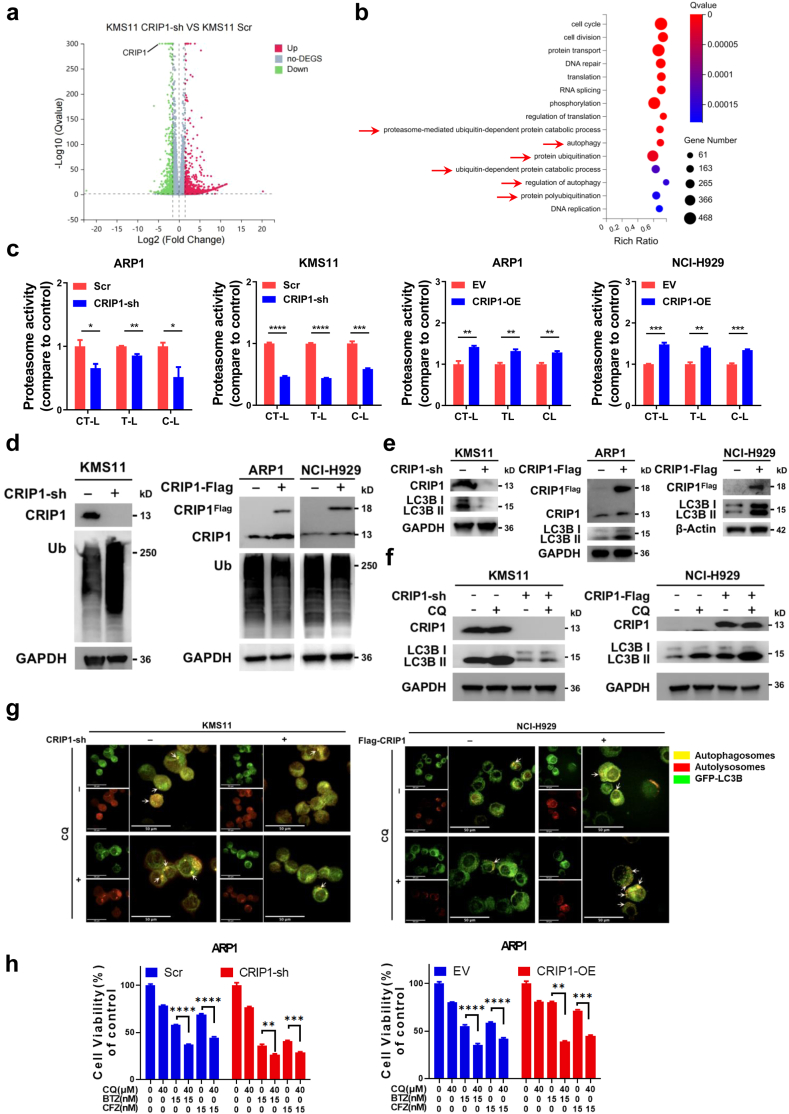
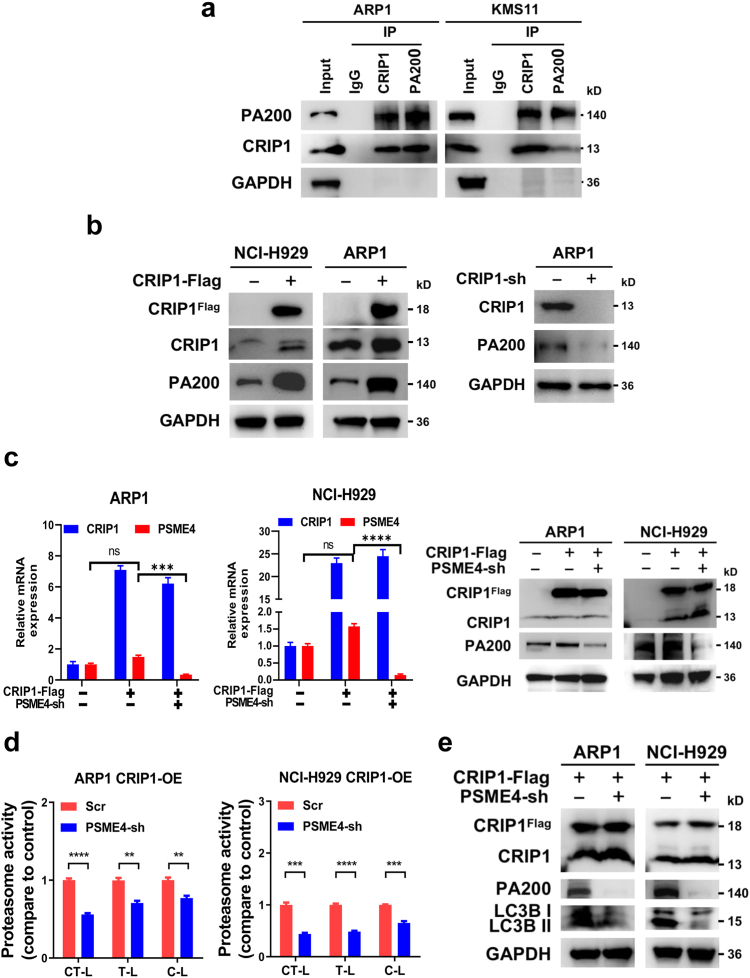
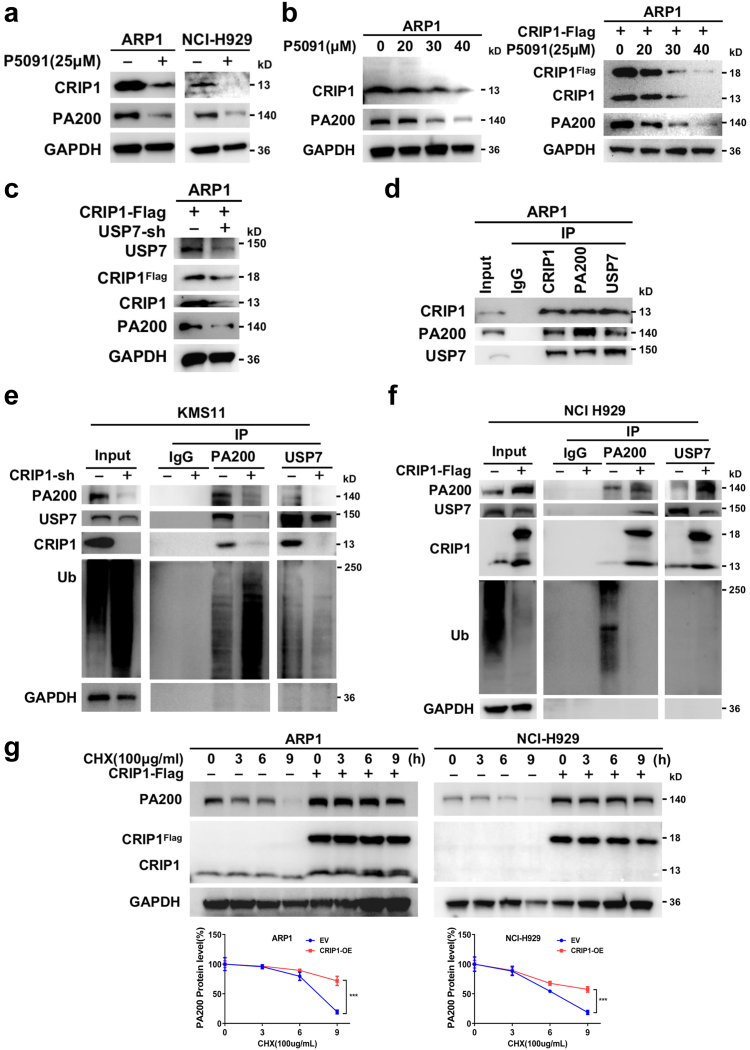
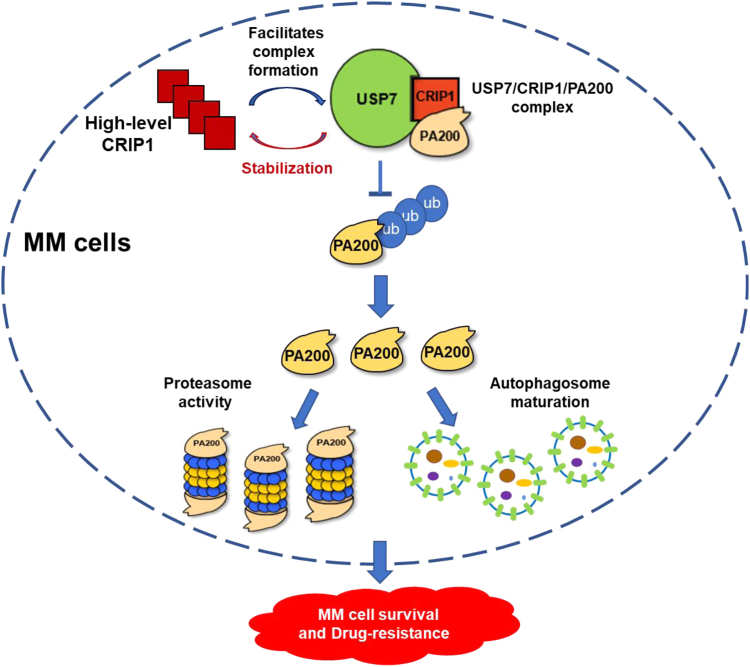
Findings: High CRIP1 expression was associated with unfavorable clinical outcomes in patients with MM and served as a biomarker for RRMM with shorter overall survival. In vitro and in vivo studies showed that CRIP1 plays a critical role in protein homeostasis via the dual regulation of the activities of proteasome and autophagy in MM cells. A combined analysis of RNA-seq, Co-IP and TAP/MS demonstrated that CRIP1 promotes proteasome inhibitors resistance in MM cells by simultaneously binding to de-ubiquitinase USP7 and proteasome coactivator PA200. CRIP1 promoted proteasome activity and autophagosome maturation by facilitating the dequbiquitination and stabilization of PA200.
Interpretation: Our findings clarified the pivotal roles of the CRIP1/USP7/PA200 complex in ubiquitin-dependent proteasome degradation and autophagy maturation involved in the pathogenesis of MM.
Funding: A full list of funding sources can be found in the acknowledgements section.
Keywords: Autophagy; CRIP1; Multiple myeloma; PIs resistance; Proteasome degradation.
Copyright © 2024 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors have declared that no conflicts of interest exist.
Figures
References
-
- van de Donk N., Pawlyn C., Yong K.L. Multiple myeloma. Lancet. 2021;397(10272):410–427. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
