

The chromatin landscape of healthy and injured cell types in the human kidney
- PMID: 38199997
- PMCID: PMC10781985
- DOI: 10.1038/s41467-023-44467-6
The chromatin landscape of healthy and injured cell types in the human kidney
Abstract
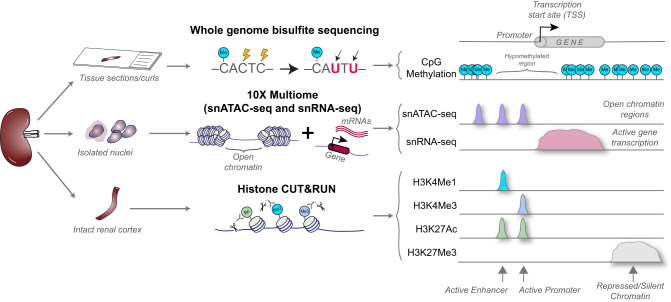
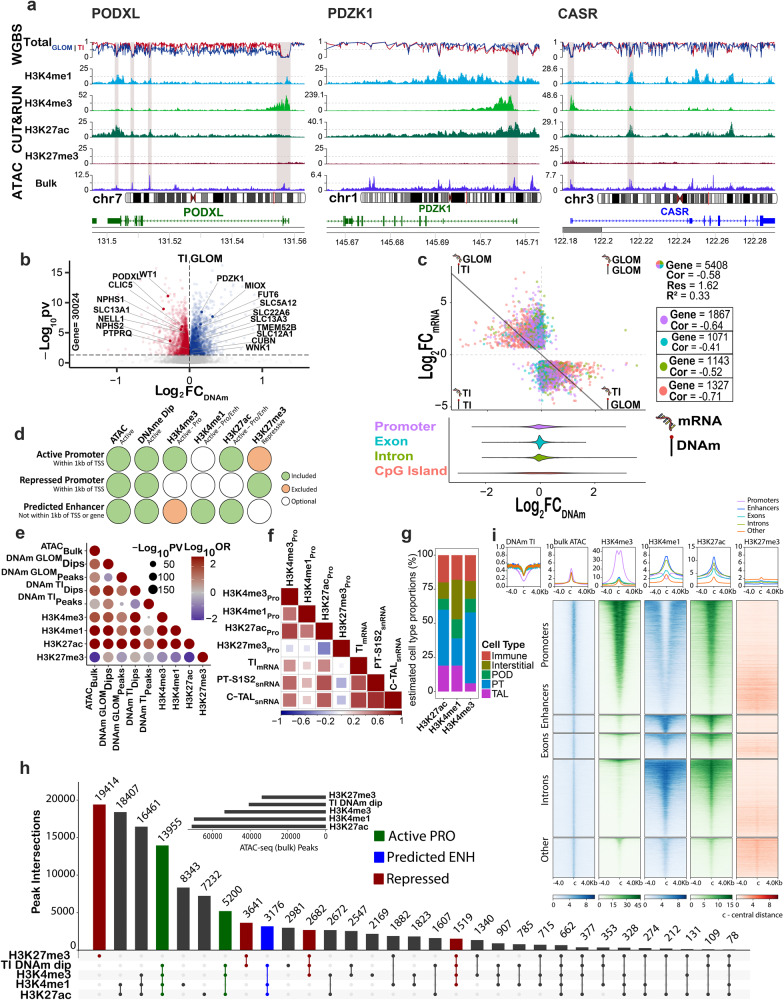
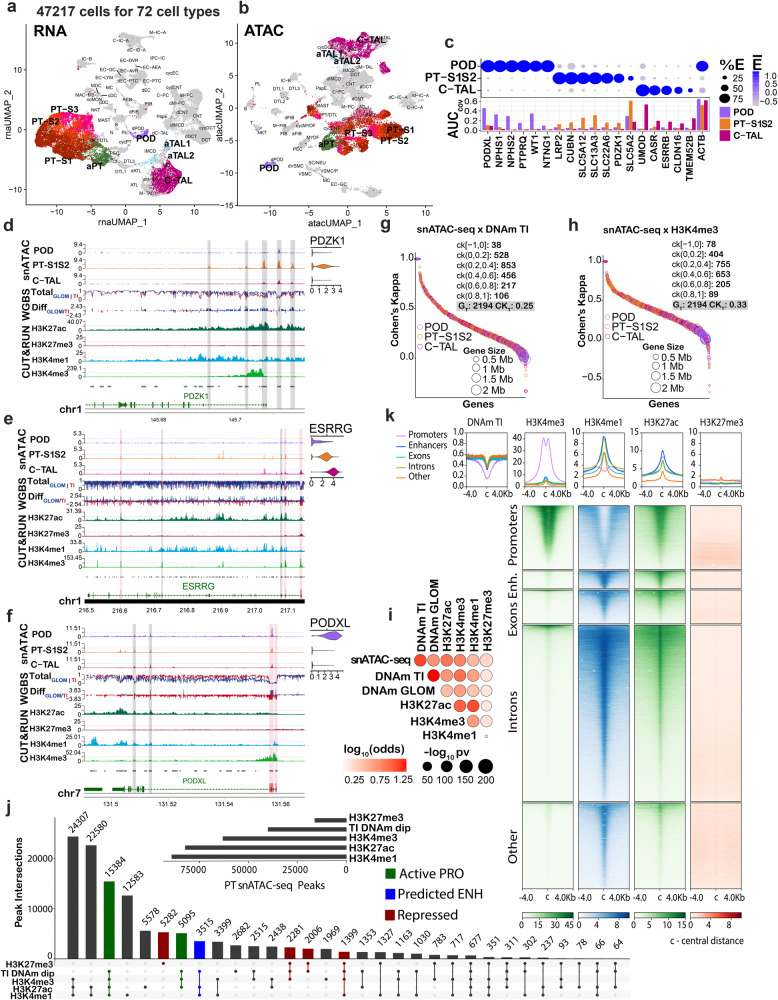
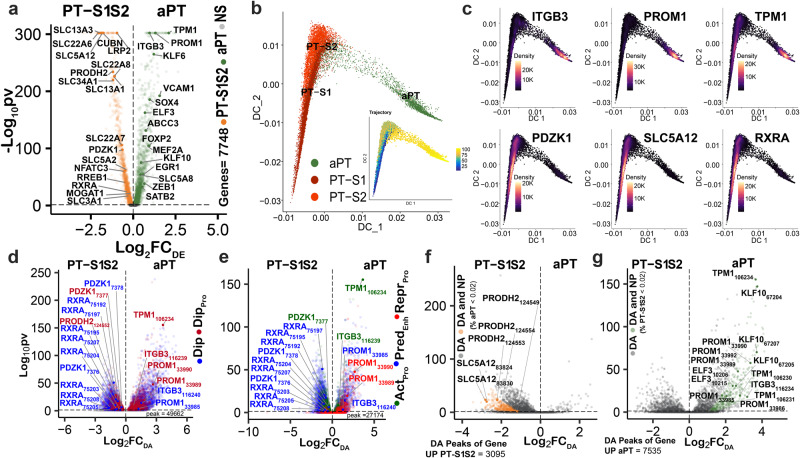
There is a need to define regions of gene activation or repression that control human kidney cells in states of health, injury, and repair to understand the molecular pathogenesis of kidney disease and design therapeutic strategies. Comprehensive integration of gene expression with epigenetic features that define regulatory elements remains a significant challenge. We measure dual single nucleus RNA expression and chromatin accessibility, DNA methylation, and H3K27ac, H3K4me1, H3K4me3, and H3K27me3 histone modifications to decipher the chromatin landscape and gene regulation of the kidney in reference and adaptive injury states. We establish a spatially-anchored epigenomic atlas to define the kidney's active, silent, and regulatory accessible chromatin regions across the genome. Using this atlas, we note distinct control of adaptive injury in different epithelial cell types. A proximal tubule cell transcription factor network of ELF3, KLF6, and KLF10 regulates the transition between health and injury, while in thick ascending limb cells this transition is regulated by NR2F1. Further, combined perturbation of ELF3, KLF6, and KLF10 distinguishes two adaptive proximal tubular cell subtypes, one of which manifested a repair trajectory after knockout. This atlas will serve as a foundation to facilitate targeted cell-specific therapeutics by reprogramming gene regulatory networks.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
The chromatin landscape of healthy and injured cell types in the human kidney.bioRxiv [Preprint]. 2023 Jun 8:2023.06.07.543965. doi: 10.1101/2023.06.07.543965. bioRxiv. 2023. Update in: Nat Commun. 2024 Jan 10;15(1):433. doi: 10.1038/s41467-023-44467-6. PMID: 37333123 Free PMC article. Updated. Preprint.
References
-
- Kellum, J. A. et al. Acute kidney injury. Nat. Rev. Dis. Primers7, 52 (2021). - PubMed
MeSH terms
Substances
Grants and funding
- U01 DK114866/DK/NIDDK NIH HHS/United States
- U01 DK133097/DK/NIDDK NIH HHS/United States
- U01 DK133090/DK/NIDDK NIH HHS/United States
- S10 OD026880/OD/NIH HHS/United States
- U01 DK114908/DK/NIDDK NIH HHS/United States
- U01 DK133095/DK/NIDDK NIH HHS/United States
- U01 DK133081/DK/NIDDK NIH HHS/United States
- U01 DK114907/DK/NIDDK NIH HHS/United States
- UH3 DK114933/DK/NIDDK NIH HHS/United States
- U24 DK114886/DK/NIDDK NIH HHS/United States
- U01 DK133766/DK/NIDDK NIH HHS/United States
- U01 DK114923/DK/NIDDK NIH HHS/United States
- U01 DK133113/DK/NIDDK NIH HHS/United States
- P50 DK133943/DK/NIDDK NIH HHS/United States
- R01 AT011463/AT/NCCIH NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- R01 DK129879/DK/NIDDK NIH HHS/United States
- U01 DK133768/DK/NIDDK NIH HHS/United States
- UH3 DK114861/DK/NIDDK NIH HHS/United States
- U01 DK133092/DK/NIDDK NIH HHS/United States
- U01 DK114933/DK/NIDDK NIH HHS/United States
- P30 DK036836/DK/NIDDK NIH HHS/United States
- UG3 DK114861/DK/NIDDK NIH HHS/United States
- U54 DK137328/DK/NIDDK NIH HHS/United States
- S10 OD030463/OD/NIH HHS/United States
- U01 DK133091/DK/NIDDK NIH HHS/United States
- U01 DK133093/DK/NIDDK NIH HHS/United States
- U54 DK134301/DK/NIDDK NIH HHS/United States
- U01 DK114920/DK/NIDDK NIH HHS/United States
