

Cryo-EM structure and B-factor refinement with ensemble representation
- PMID: 38200043
- PMCID: PMC10781738
- DOI: 10.1038/s41467-023-44593-1
Cryo-EM structure and B-factor refinement with ensemble representation
Abstract
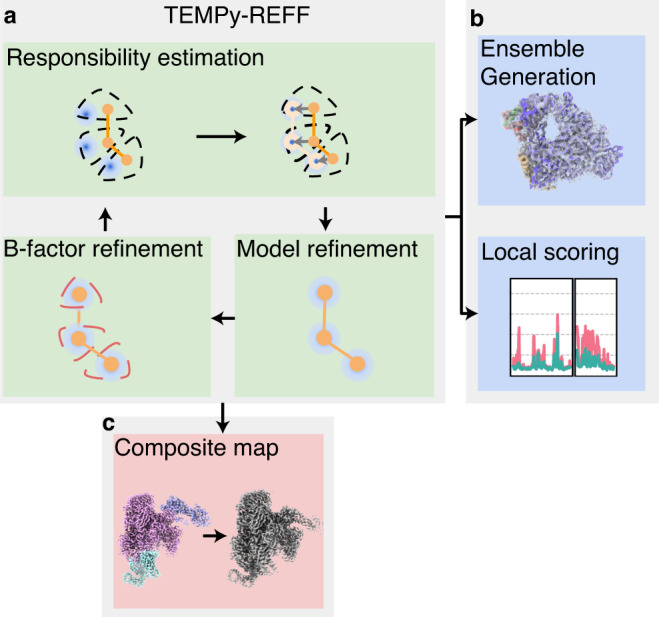
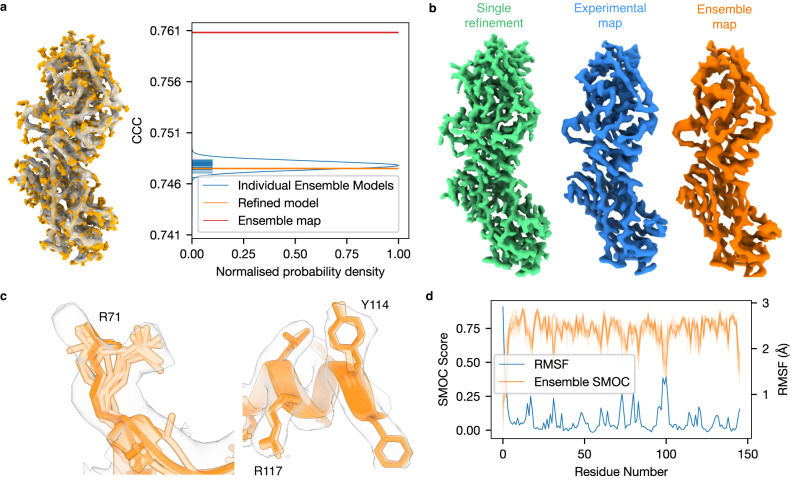
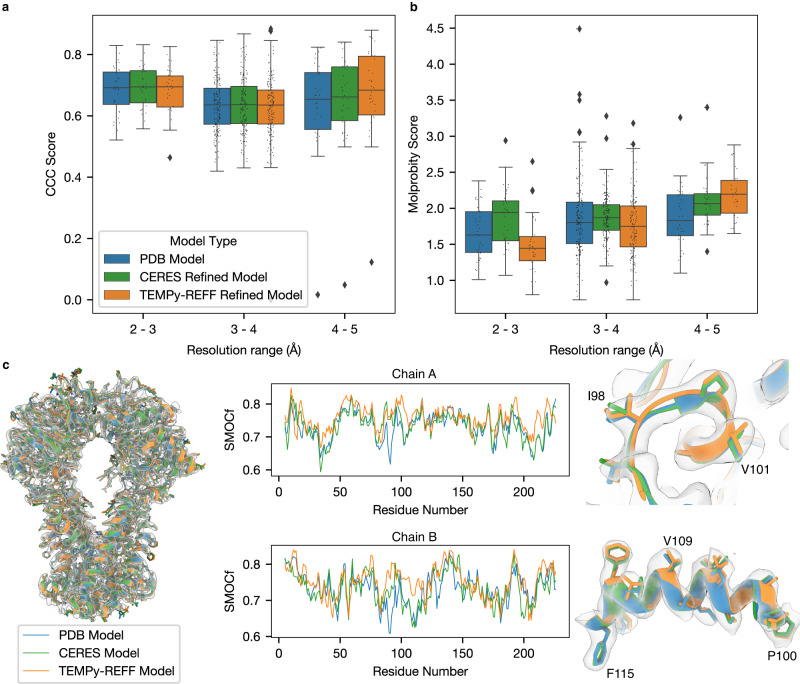
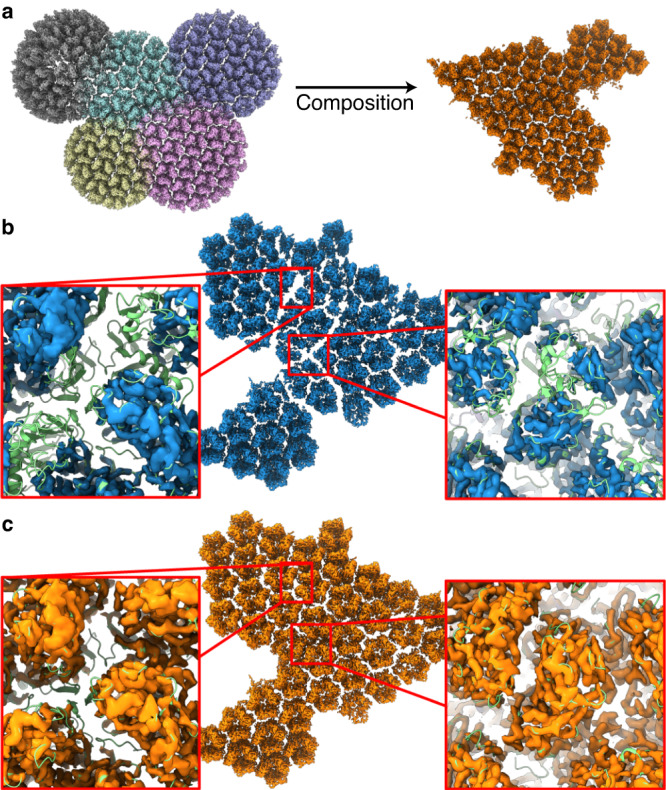
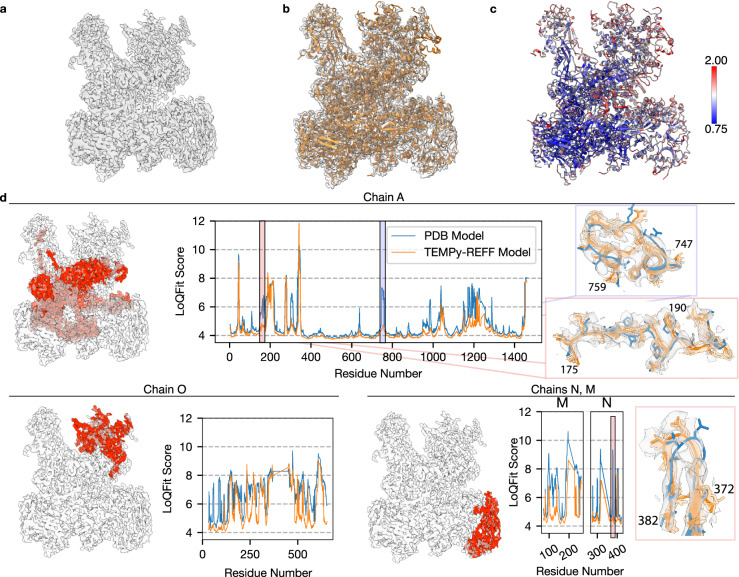
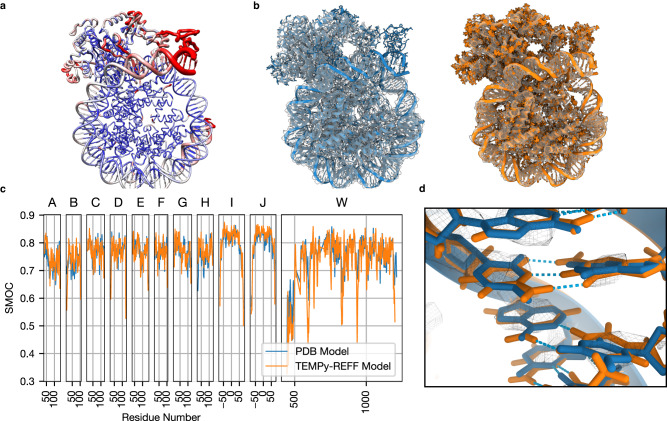
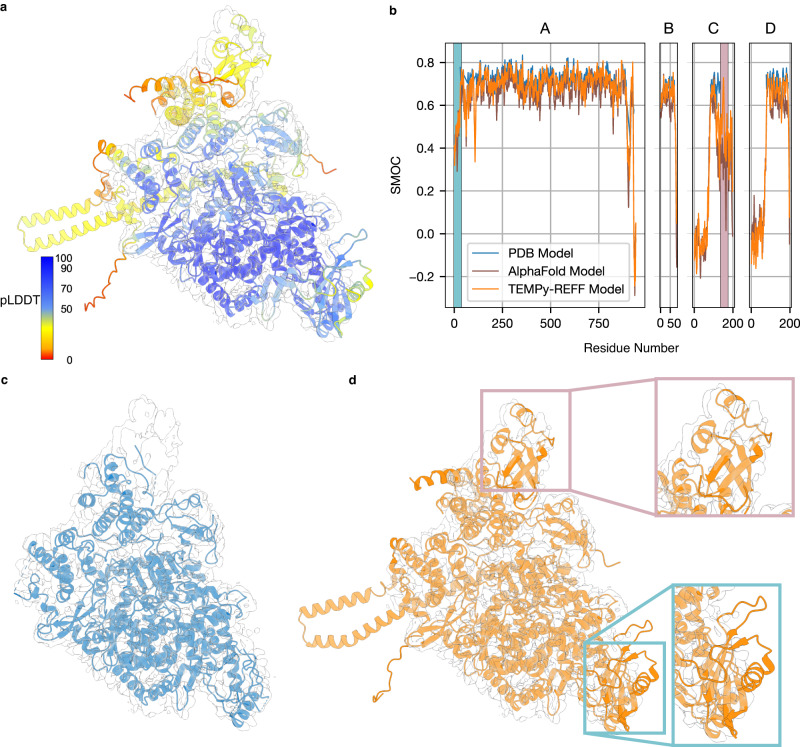
Cryo-EM experiments produce images of macromolecular assemblies that are combined to produce three-dimensional density maps. Typically, atomic models of the constituent molecules are fitted into these maps, followed by a density-guided refinement. We introduce TEMPy-ReFF, a method for atomic structure refinement in cryo-EM density maps. Our method represents atomic positions as components of a Gaussian mixture model, utilising their variances as B-factors, which are used to derive an ensemble description. Extensively tested on a substantial dataset of 229 cryo-EM maps from EMDB ranging in resolution from 2.1-4.9 Å with corresponding PDB and CERES atomic models, our results demonstrate that TEMPy-ReFF ensembles provide a superior representation of cryo-EM maps. On a single-model basis, it performs similarly to the CERES re-refinement protocol, although there are cases where it provides a better fit to the map. Furthermore, our method enables the creation of composite maps free of boundary artefacts. TEMPy-ReFF is useful for better interpretation of flexible structures, such as those involving RNA, DNA or ligands.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta.Elife. 2016 Sep 26;5:e17219. doi: 10.7554/eLife.17219. Elife. 2016. PMID: 27669148 Free PMC article.
-
A New Protocol for Atomic-Level Protein Structure Modeling and Refinement Using Low-to-Medium Resolution Cryo-EM Density Maps.J Mol Biol. 2020 Sep 4;432(19):5365-5377. doi: 10.1016/j.jmb.2020.07.027. Epub 2020 Aug 6. J Mol Biol. 2020. PMID: 32771523 Free PMC article.
-
Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement.J Chem Inf Model. 2020 May 26;60(5):2522-2532. doi: 10.1021/acs.jcim.9b00932. Epub 2019 Dec 24. J Chem Inf Model. 2020. PMID: 31872764 Free PMC article.
-
Refinement of Atomic Structures Against cryo-EM Maps.Methods Enzymol. 2016;579:277-305. doi: 10.1016/bs.mie.2016.05.033. Epub 2016 Jun 24. Methods Enzymol. 2016. PMID: 27572731 Review.
-
Evolution of standardization and dissemination of cryo-EM structures and data jointly by the community, PDB, and EMDB.J Biol Chem. 2021 Jan-Jun;296:100560. doi: 10.1016/j.jbc.2021.100560. Epub 2021 Mar 18. J Biol Chem. 2021. PMID: 33744287 Free PMC article. Review.
Cited by
-
Accurate model and ensemble refinement using cryo-electron microscopy maps and Bayesian inference.PLoS Comput Biol. 2024 Jul 15;20(7):e1012180. doi: 10.1371/journal.pcbi.1012180. eCollection 2024 Jul. PLoS Comput Biol. 2024. PMID: 39008528 Free PMC article.
-
Frontiers in integrative structural modeling of macromolecular assemblies.QRB Discov. 2025 Jan 22;6:e3. doi: 10.1017/qrd.2024.15. eCollection 2025. QRB Discov. 2025. PMID: 39944881 Free PMC article. Review.
-
CASP15 cryo-EM protein and RNA targets: Refinement and analysis using experimental maps.Proteins. 2023 Dec;91(12):1935-1951. doi: 10.1002/prot.26644. Proteins. 2023. PMID: 37994556 Free PMC article.
-
How structural biology has changed our understanding of icosahedral viruses.J Virol. 2024 Oct 22;98(10):e0111123. doi: 10.1128/jvi.01111-23. Epub 2024 Sep 18. J Virol. 2024. PMID: 39291975 Free PMC article. Review.
-
as a reliability measure for protein, nucleic acid, and small molecule atomic coordinate models derived from 3DEM density maps.bioRxiv [Preprint]. 2025 Jan 15:2025.01.14.633006. doi: 10.1101/2025.01.14.633006. bioRxiv. 2025. Update in: Acta Crystallogr D Struct Biol. 2025 Aug 1;81(Pt 8):410-422. doi: 10.1107/S2059798325005923. PMID: 39868161 Free PMC article. Updated. Preprint.
References
-
- van Zundert GydoCP, Bijvoet Center for Biomolecular Research, Faculty of Science-Chemistry, Utrecht University, Utrecht, the Netherlands. Bonvin AlexandreMJJ. Fast and sensitive rigid-body fitting into cryo-EM density maps with PowerFit. AIMS Biophys. 2015;2:73–87. doi: 10.3934/biophy.2015.2.73. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
