

Profiling the heterogeneity of colorectal cancer consensus molecular subtypes using spatial transcriptomics
- PMID: 38200223
- PMCID: PMC10781769
- DOI: 10.1038/s41698-023-00488-4
Profiling the heterogeneity of colorectal cancer consensus molecular subtypes using spatial transcriptomics
Abstract
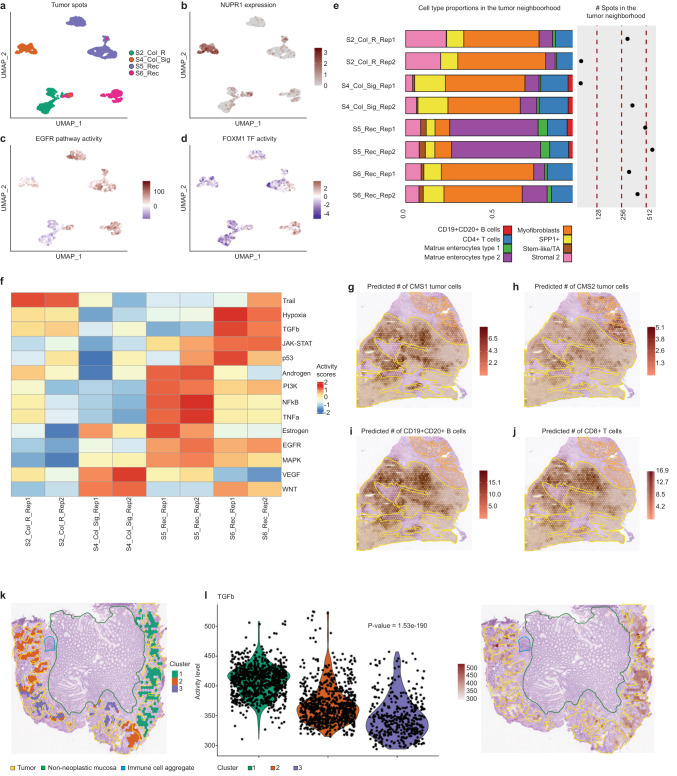
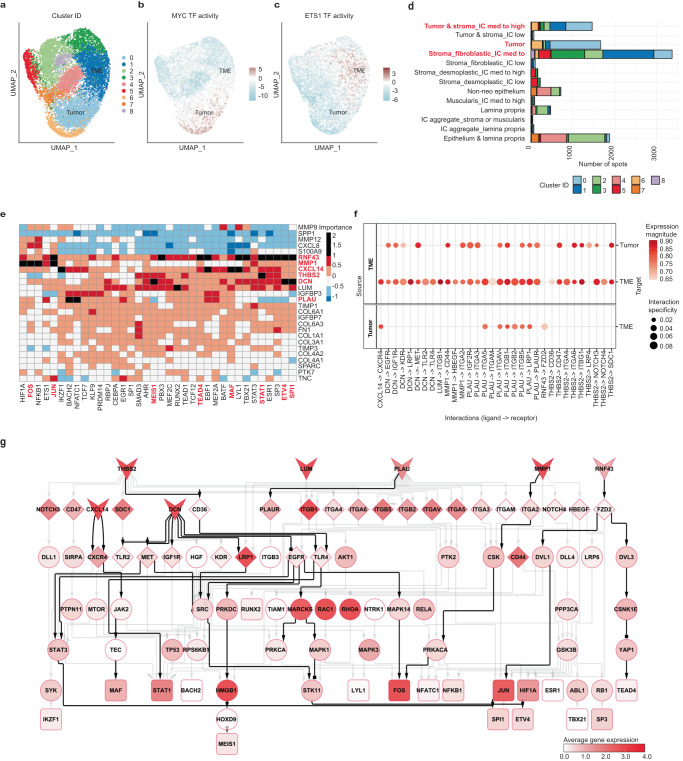
The consensus molecular subtypes (CMS) of colorectal cancer (CRC) is the most widely-used gene expression-based classification and has contributed to a better understanding of disease heterogeneity and prognosis. Nevertheless, CMS intratumoral heterogeneity restricts its clinical application, stressing the necessity of further characterizing the composition and architecture of CRC. Here, we used Spatial Transcriptomics (ST) in combination with single-cell RNA sequencing (scRNA-seq) to decipher the spatially resolved cellular and molecular composition of CRC. In addition to mapping the intratumoral heterogeneity of CMS and their microenvironment, we identified cell communication events in the tumor-stroma interface of CMS2 carcinomas. This includes tumor growth-inhibiting as well as -activating signals, such as the potential regulation of the ETV4 transcriptional activity by DCN or the PLAU-PLAUR ligand-receptor interaction. Our study illustrates the potential of ST to resolve CRC molecular heterogeneity and thereby help advance personalized therapy.
© 2024. The Author(s).
Conflict of interest statement
A.V., B.A., E.G., N.G., M.R., S.B., I.W., B.P., L.V., E.Y., M.B., M.S., N.K., B.J., P.S., T.B. and K.H. are currently employed by F. Hoffmann-La Roche Ltd. A.J.L. and D.T. were previously employed by F. Hoffmann-La Roche Ltd. A.J.L. is currently employed by Idorsia Pharmaceuticals Ltd. D.T. is currently employed by University of Bern. A.L. is currently employed by Genentech, Inc. M.D.T. was previously employed by Genentech, Inc and is currently employed by Gilead Sciences, Inc. J.S.R. has received funding from GSK and Sanofi and fees from Travere Therapeutics and Astex Pharmaceuticals. The authors declare that they have no other competing interests.
Figures




References
LinkOut - more resources
Full Text Sources
Miscellaneous
