

Insights for precision oncology from the integration of genomic and clinical data of 13,880 tumors from the 100,000 Genomes Cancer Programme
- PMID: 38200255
- PMCID: PMC10803271
- DOI: 10.1038/s41591-023-02682-0
Insights for precision oncology from the integration of genomic and clinical data of 13,880 tumors from the 100,000 Genomes Cancer Programme
Abstract
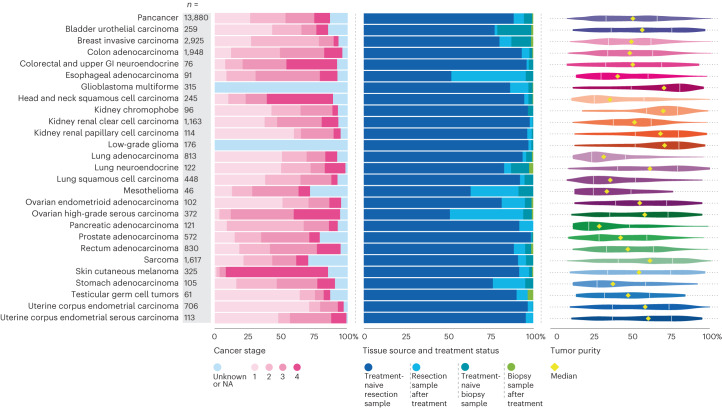
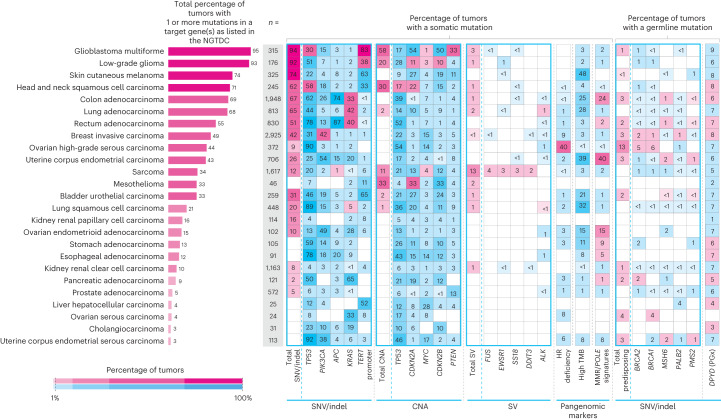
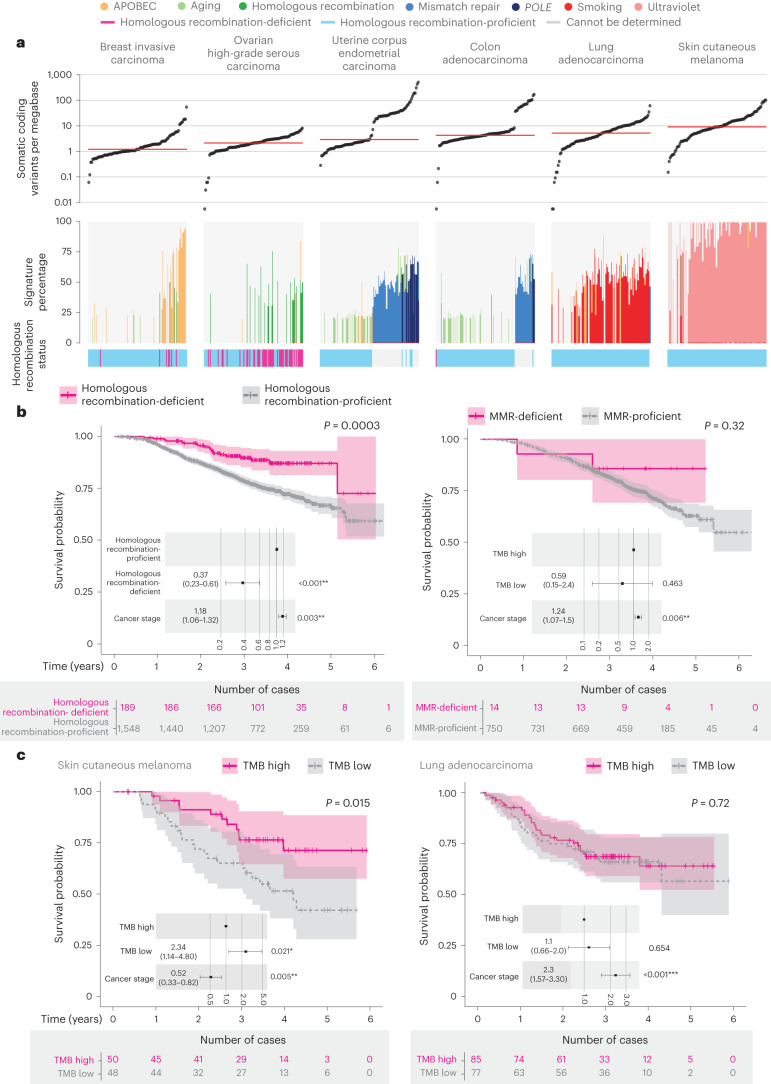
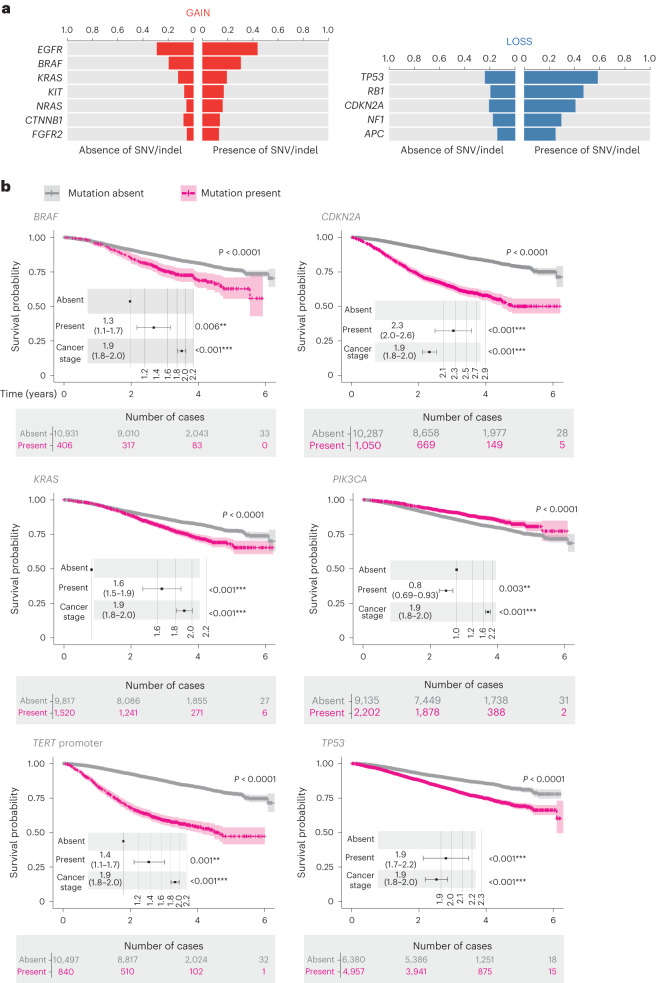
The Cancer Programme of the 100,000 Genomes Project was an initiative to provide whole-genome sequencing (WGS) for patients with cancer, evaluating opportunities for precision cancer care within the UK National Healthcare System (NHS). Genomics England, alongside NHS England, analyzed WGS data from 13,880 solid tumors spanning 33 cancer types, integrating genomic data with real-world treatment and outcome data, within a secure Research Environment. Incidence of somatic mutations in genes recommended for standard-of-care testing varied across cancer types. For instance, in glioblastoma multiforme, small variants were present in 94% of cases and copy number aberrations in at least one gene in 58% of cases, while sarcoma demonstrated the highest occurrence of actionable structural variants (13%). Homologous recombination deficiency was identified in 40% of high-grade serous ovarian cancer cases with 30% linked to pathogenic germline variants, highlighting the value of combined somatic and germline analysis. The linkage of WGS and longitudinal life course clinical data allowed the assessment of treatment outcomes for patients stratified according to pangenomic markers. Our findings demonstrate the utility of linking genomic and real-world clinical data to enable survival analysis to identify cancer genes that affect prognosis and advance our understanding of how cancer genomics impacts patient outcomes.
© 2024. The Author(s).
Conflict of interest statement
Genomics England is a company wholly owned by the UK DHSC and was created in 2013 to introduce WGS into healthcare in conjunction with NHS England. All authors affiliated with Genomics England (A. Sosinsky, J.A., C.T., S. Henderson, L.J., A.H., P.A., G.C., J.M., S.W., K.B., D.P., M.B.P., N.V., A.R.-M., D.P.-G., J.L., J.P., A. Siddiq, T.Z., T.C., O.Y., T.F., A.R., M.C. and N.M.) are, or were, salaried by or seconded to Genomics England. D.B. and C.K. are full-time employees and shareholders of Illumina. A.H. has received speaker fees from Gilead, Roche, Pfizer, Jazz, AbbVie, Incyte and Astellas. N.M. has provided consulting and advisory support for Pfizer, Guardant, Seagen and Janssen, and received speaker fees from Novartis, Pfizer and Servier outside of the submitted work. The remaining authors declare no competing interests.
Figures




Comment in
-
The grand challenge of moving cancer whole-genome sequencing into the clinic.Nat Med. 2024 Jan;30(1):39-40. doi: 10.1038/s41591-023-02697-7. Nat Med. 2024. PMID: 38200256 No abstract available.
References
-
- Cancer Incidence Statistics. Cancer Research UKwww.cancerresearchuk.org/health-professional/cancer-statistics/incidence (undated).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
