

Elevated genetic risk for multiple sclerosis emerged in steppe pastoralist populations
- PMID: 38200296
- PMCID: PMC10781639
- DOI: 10.1038/s41586-023-06618-z
Elevated genetic risk for multiple sclerosis emerged in steppe pastoralist populations
Abstract
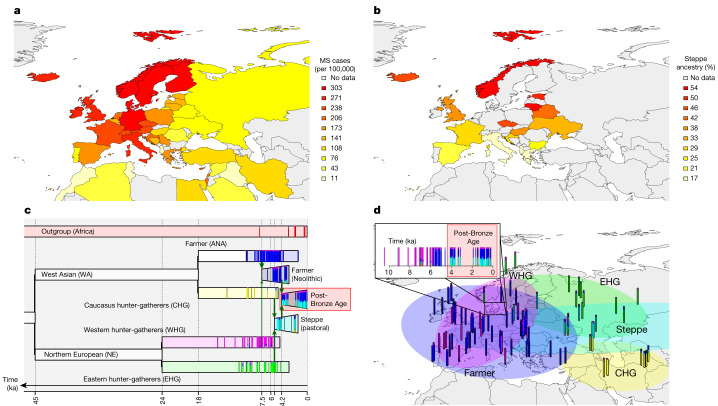
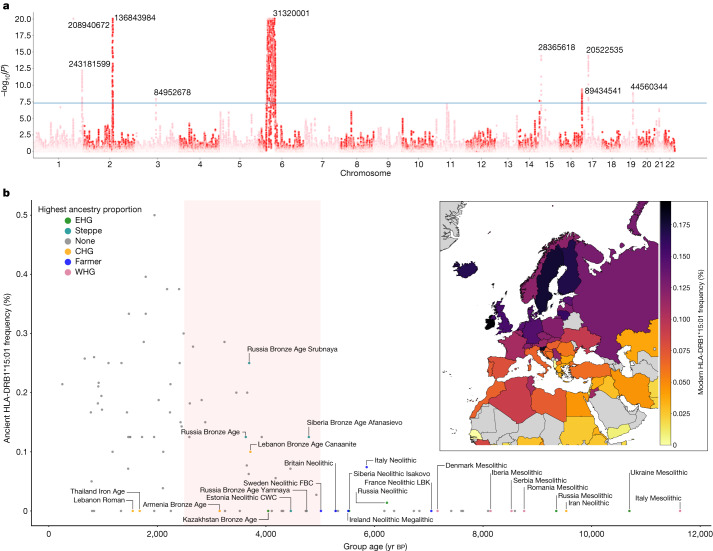
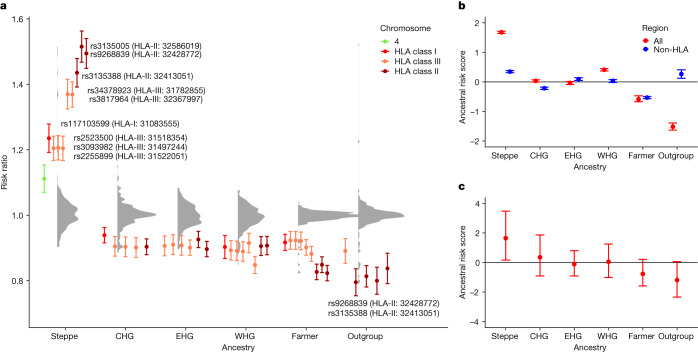
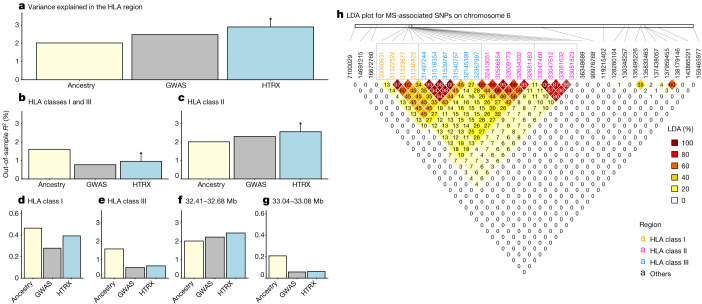
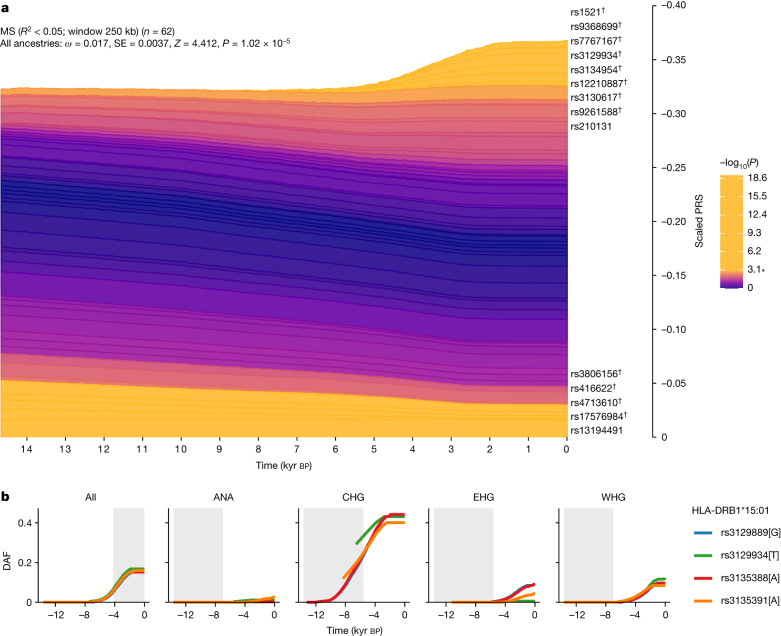
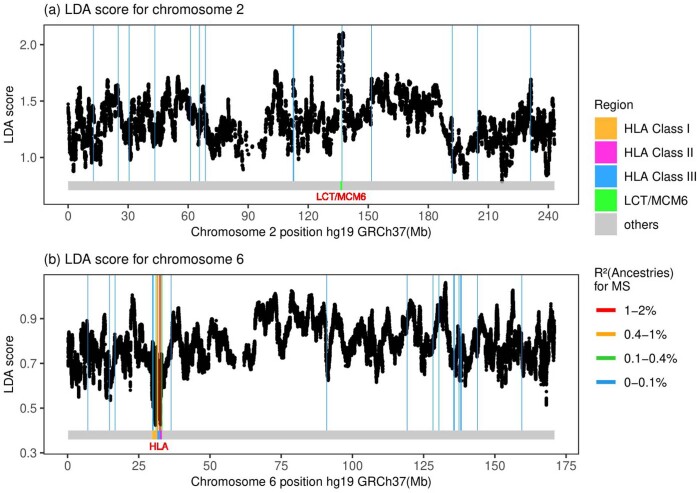
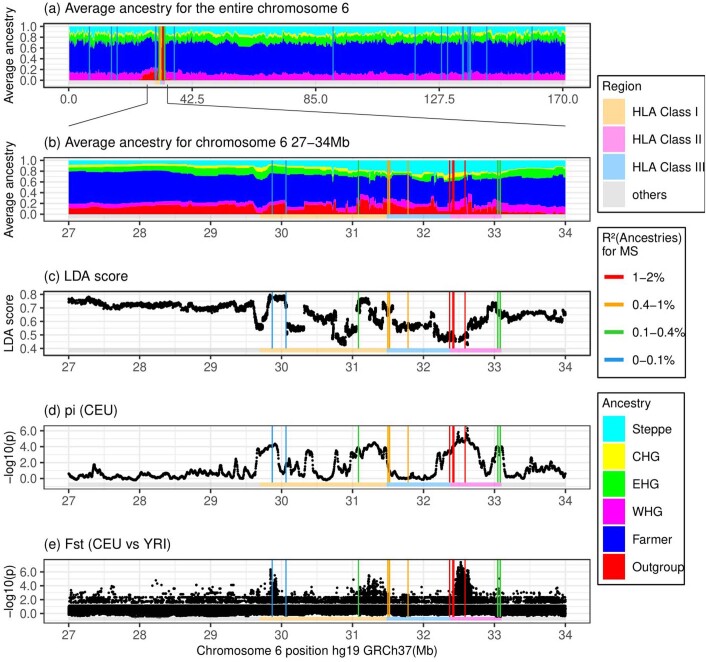
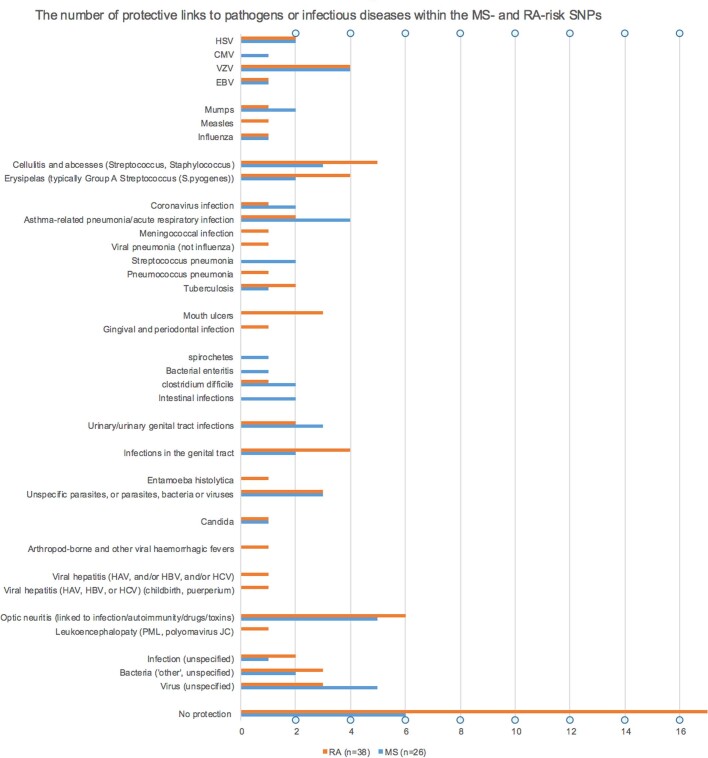
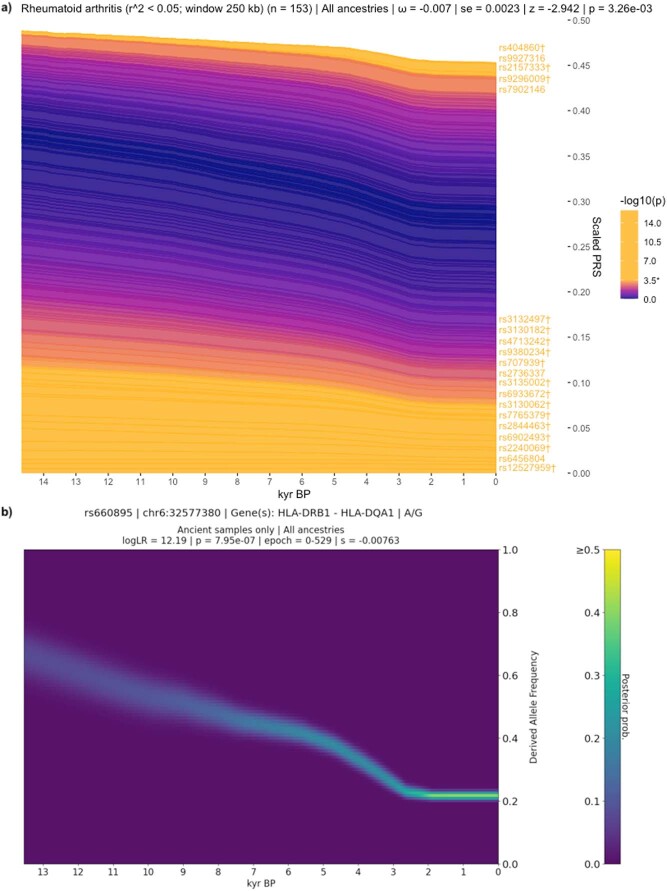
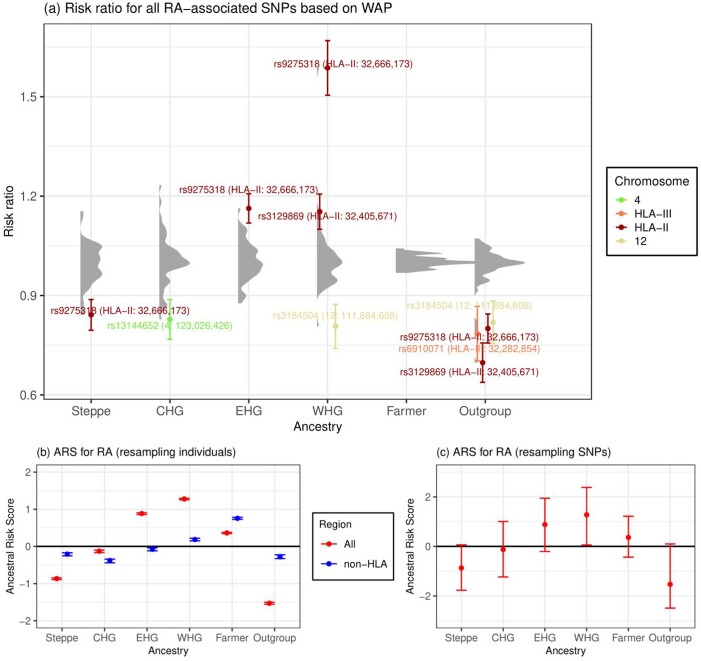
Multiple sclerosis (MS) is a neuro-inflammatory and neurodegenerative disease that is most prevalent in Northern Europe. Although it is known that inherited risk for MS is located within or in close proximity to immune-related genes, it is unknown when, where and how this genetic risk originated1. Here, by using a large ancient genome dataset from the Mesolithic period to the Bronze Age2, along with new Medieval and post-Medieval genomes, we show that the genetic risk for MS rose among pastoralists from the Pontic steppe and was brought into Europe by the Yamnaya-related migration approximately 5,000 years ago. We further show that these MS-associated immunogenetic variants underwent positive selection both within the steppe population and later in Europe, probably driven by pathogenic challenges coinciding with changes in diet, lifestyle and population density. This study highlights the critical importance of the Neolithic period and Bronze Age as determinants of modern immune responses and their subsequent effect on the risk of developing MS in a changing environment.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Attfield, K. E., Jensen, L. T., Kaufmann, M., Friese, M. A. & Fugger, L. The immunology of multiple sclerosis. Nat. Rev. Immunol.10.1038/s41577-022-00718-z (2022). - PubMed
