

Targeting the SphK1/S1P/PFKFB3 axis suppresses hepatocellular carcinoma progression by disrupting glycolytic energy supply that drives tumor angiogenesis
- PMID: 38200582
- PMCID: PMC10782643
- DOI: 10.1186/s12967-023-04830-z
Targeting the SphK1/S1P/PFKFB3 axis suppresses hepatocellular carcinoma progression by disrupting glycolytic energy supply that drives tumor angiogenesis
Abstract
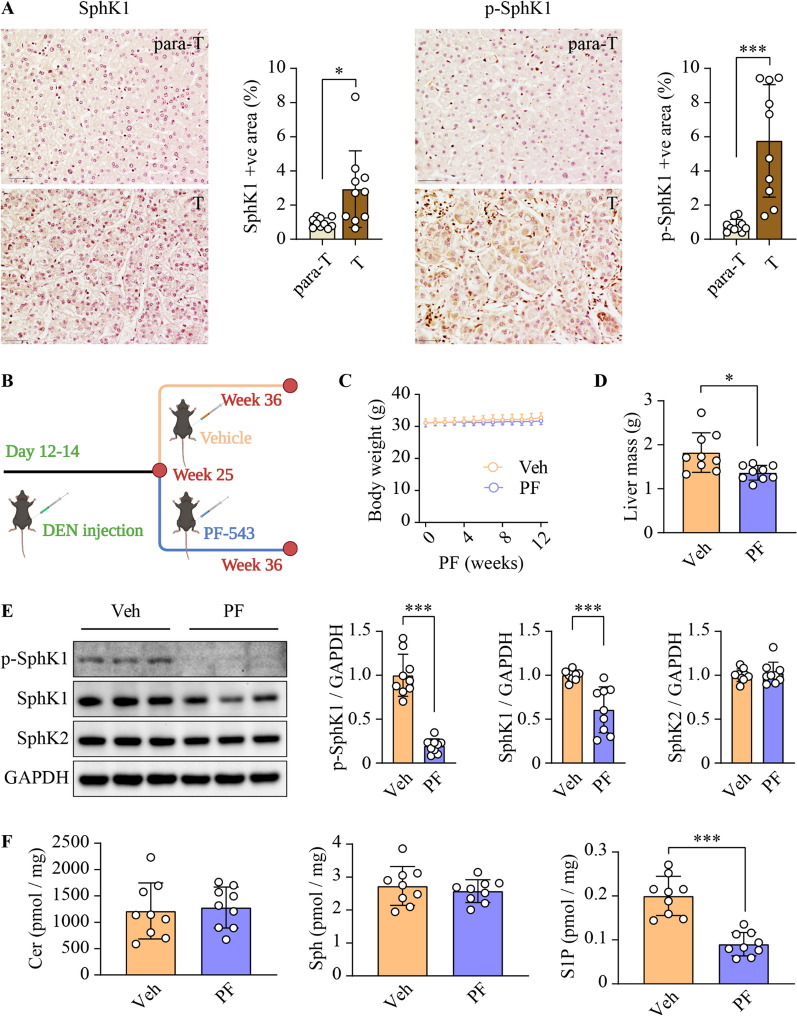
Background: Hepatocellular carcinoma (HCC) remains a leading life-threatening health challenge worldwide, with pressing needs for novel therapeutic strategies. Sphingosine kinase 1 (SphK1), a well-established pro-cancer enzyme, is aberrantly overexpressed in a multitude of malignancies, including HCC. Our previous research has shown that genetic ablation of Sphk1 mitigates HCC progression in mice. Therefore, the development of PF-543, a highly selective SphK1 inhibitor, opens a new avenue for HCC treatment. However, the anti-cancer efficacy of PF-543 has not yet been investigated in primary cancer models in vivo, thereby limiting its further translation.
Methods: Building upon the identification of the active form of SphK1 as a viable therapeutic target in human HCC specimens, we assessed the capacity of PF-543 in suppressing tumor progression using a diethylnitrosamine-induced mouse model of primary HCC. We further delineated its underlying mechanisms in both HCC and endothelial cells. Key findings were validated in Sphk1 knockout mice and lentiviral-mediated SphK1 knockdown cells.
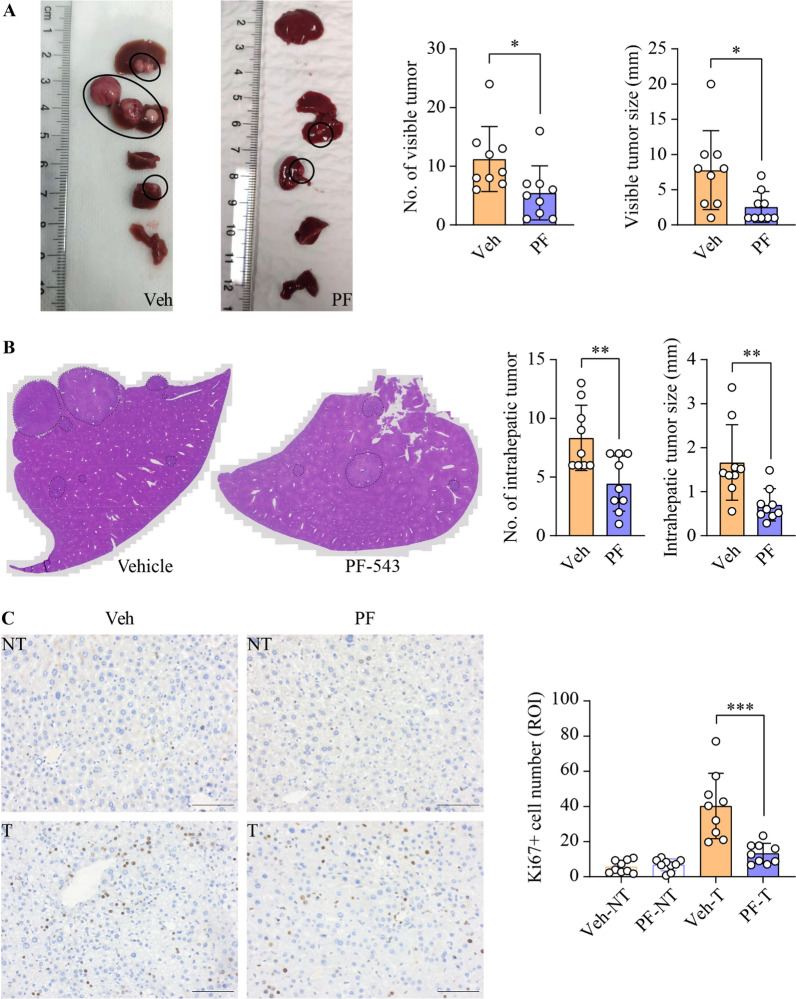
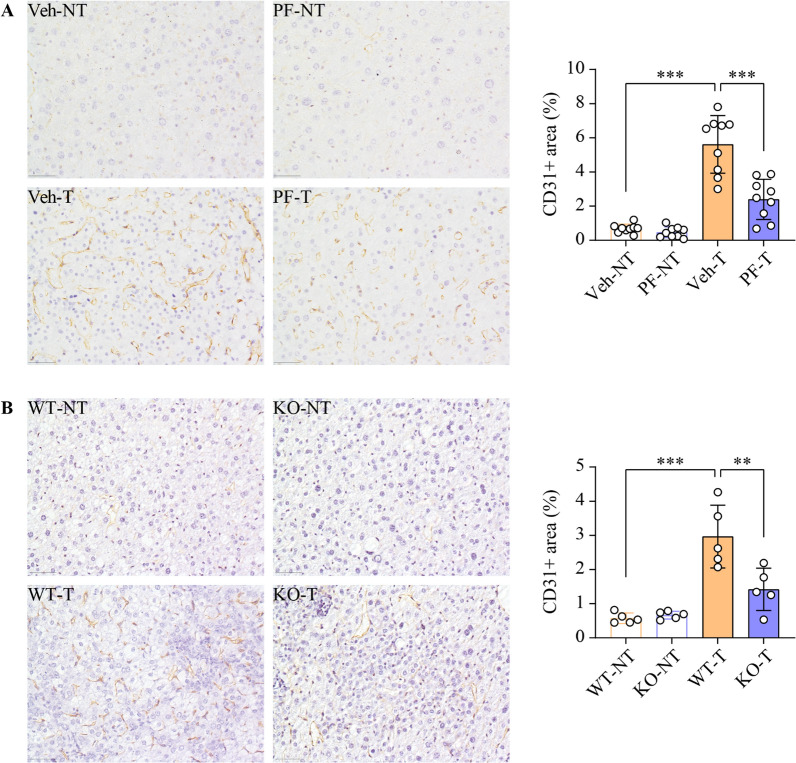
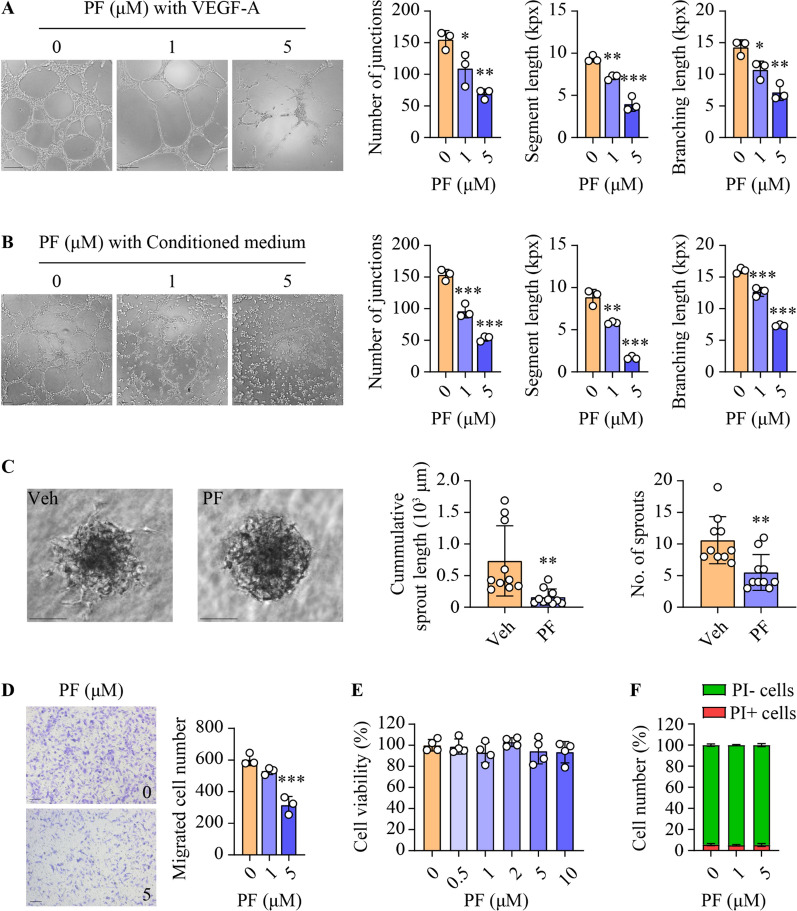
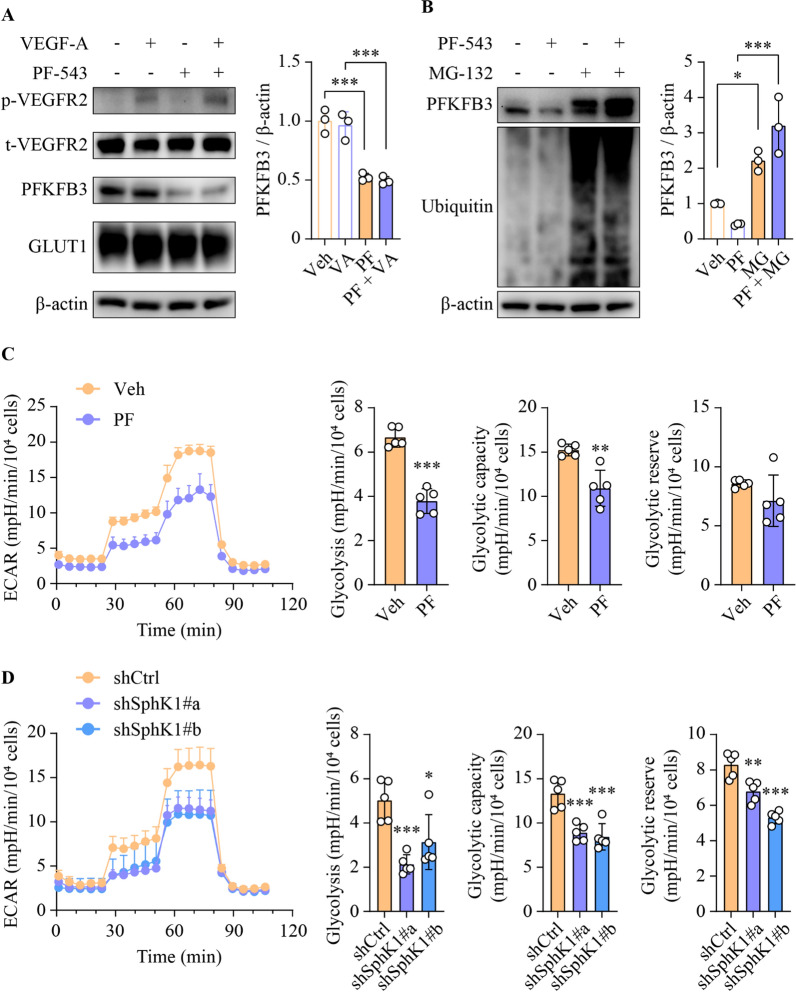
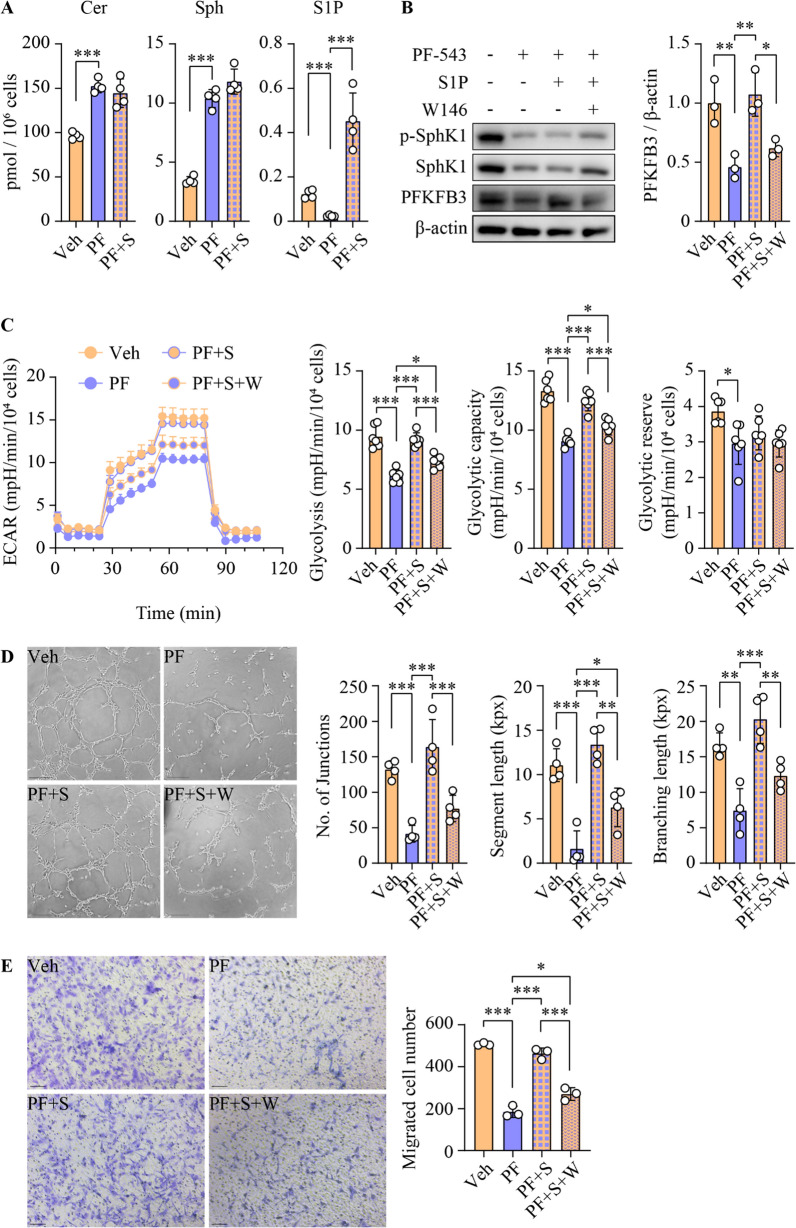
Results: SphK1 activity was found to be elevated in human HCC tissues. Administration of PF-543 effectively abrogated hepatic SphK1 activity and significantly suppressed HCC progression in diethylnitrosamine-treated mice. The primary mechanism of action was through the inhibition of tumor neovascularization, as PF-543 disrupted endothelial cell angiogenesis even in a pro-angiogenic milieu. Mechanistically, PF-543 induced proteasomal degradation of the critical glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, thus restricting the energy supply essential for tumor angiogenesis. These effects of PF-543 could be reversed upon S1P supplementation in an S1P receptor-dependent manner.
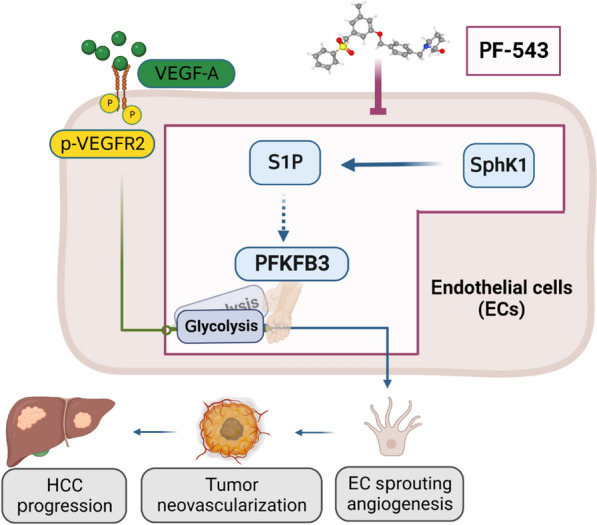
Conclusions: This study provides the first in vivo evidence supporting the potential of PF-543 as an effective anti-HCC agent. It also uncovers previously undescribed links between the pro-cancer, pro-angiogenic and pro-glycolytic roles of the SphK1/S1P/S1P receptor axis. Importantly, unlike conventional anti-HCC drugs that target individual pro-angiogenic drivers, PF-543 impairs the PFKFB3-dictated glycolytic energy engine that fuels tumor angiogenesis, representing a novel and potentially safer therapeutic strategy for HCC.
Keywords: Angiogenesis; Glycolysis; Hepatocellular carcinoma; PF-543; PFKFB3; Sphingosine kinase.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
