

Mitochondrial Targeting against Alzheimer's Disease: Lessons from Hibernation
- PMID: 38201215
- PMCID: PMC10778235
- DOI: 10.3390/cells13010012
Mitochondrial Targeting against Alzheimer's Disease: Lessons from Hibernation
Abstract
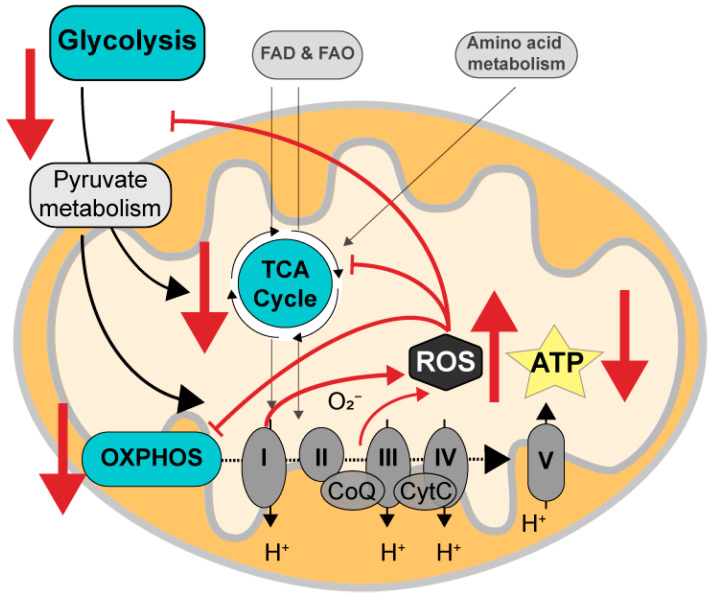
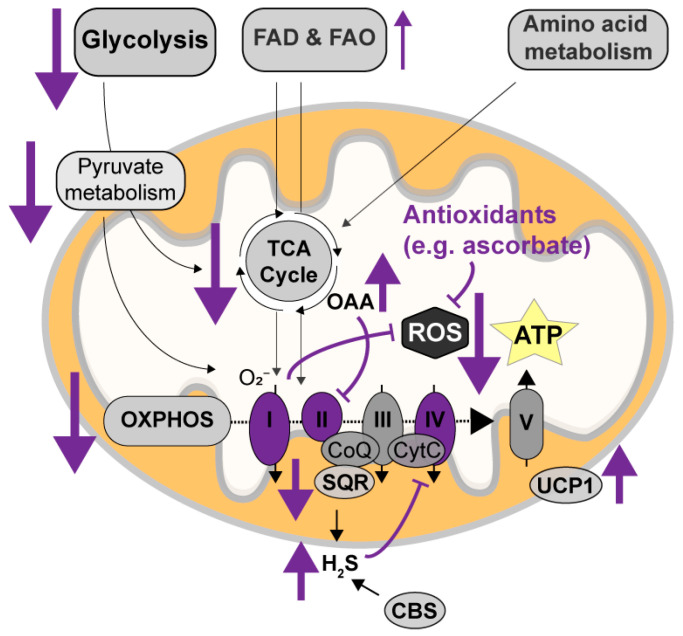
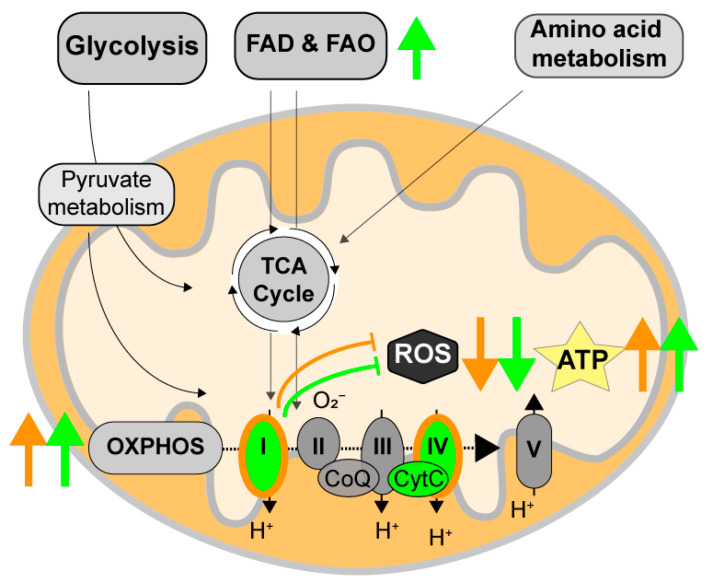
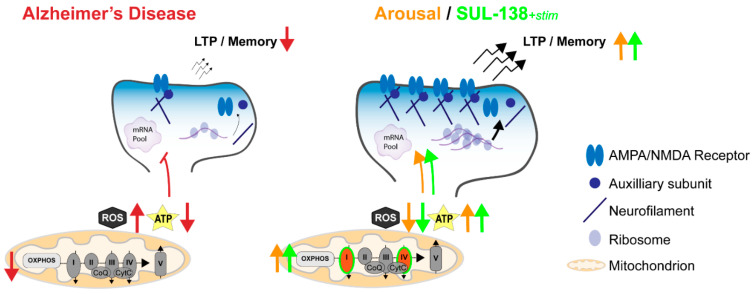
Alzheimer's disease (AD) is the most common cause of dementia worldwide and yet remains without effective therapy. Amongst the many proposed causes of AD, the mitochondrial cascade hypothesis is gaining attention. Accumulating evidence shows that mitochondrial dysfunction is a driving force behind synaptic dysfunction and cognitive decline in AD patients. However, therapies targeting the mitochondria in AD have proven unsuccessful so far, and out-of-the-box options, such as hibernation-derived mitochondrial mechanisms, may provide valuable new insights. Hibernators uniquely and rapidly alternate between suppression and re-activation of the mitochondria while maintaining a sufficient energy supply and without acquiring ROS damage. Here, we briefly give an overview of mitochondrial dysfunction in AD, how it affects synaptic function, and why mitochondrial targeting in AD has remained unsuccessful so far. We then discuss mitochondria in hibernation and daily torpor in mice, covering current advancements in hibernation-derived mitochondrial targeting strategies. We conclude with new ideas on how hibernation-derived dual mitochondrial targeting of both the ATP and ROS pathways may boost mitochondrial health and induce local synaptic protein translation to increase synaptic function and plasticity. Further exploration of these mechanisms may provide more effective treatment options for AD in the future.
Keywords: Alzheimer’s disease; SUL-138; daily torpor; hibernation-derived compound; mitochondrial dysfunction.
Conflict of interest statement
R.H.H. and C.F.d.V.M. are inventors on patent: Compounds for the treatment of Alzheimer’s Disease; patent number: WO2021118359. They do not gain financial benefits from this inventorship.
Figures
Similar articles
-
The hibernation-derived compound SUL-138 shifts the mitochondrial proteome towards fatty acid metabolism and prevents cognitive decline and amyloid plaque formation in an Alzheimer's disease mouse model.Alzheimers Res Ther. 2022 Dec 9;14(1):183. doi: 10.1186/s13195-022-01127-z. Alzheimers Res Ther. 2022. PMID: 36482297 Free PMC article.
-
Mitochondrial Dysfunction and Synaptic Transmission Failure in Alzheimer's Disease.J Alzheimers Dis. 2017;57(4):1071-1086. doi: 10.3233/JAD-160702. J Alzheimers Dis. 2017. PMID: 27662318 Free PMC article. Review.
-
Mitochondrial Dysfunction Triggers Synaptic Deficits via Activation of p38 MAP Kinase Signaling in Differentiated Alzheimer's Disease Trans-Mitochondrial Cybrid Cells.J Alzheimers Dis. 2017;59(1):223-239. doi: 10.3233/JAD-170283. J Alzheimers Dis. 2017. PMID: 28598851 Free PMC article.
-
Drug Target to Alleviate Mitochondrial Dysfunctions in Alzheimer's Disease: Recent Advances and Therapeutic Implications.Curr Neuropharmacol. 2024;22(12):1942-1959. doi: 10.2174/1570159X22666240426091311. Curr Neuropharmacol. 2024. PMID: 39234772 Free PMC article. Review.
-
Torpor enhances synaptic strength and restores memory performance in a mouse model of Alzheimer's disease.Sci Rep. 2021 Jul 29;11(1):15486. doi: 10.1038/s41598-021-94992-x. Sci Rep. 2021. PMID: 34326412 Free PMC article.
Cited by
-
The NRF2/ID2 Axis in Vascular Smooth Muscle Cells: Novel Insights into the Interplay between Vascular Calcification and Aging.Aging Dis. 2024 May 20;16(2):1120-1140. doi: 10.14336/AD.2024.0075. Aging Dis. 2024. PMID: 38916733 Free PMC article.
-
Activation of a Src-JNK pathway in unscheduled endocycling cells of the Drosophila wing disc induces a chronic wounding response.bioRxiv [Preprint]. 2025 Mar 13:2025.03.12.642788. doi: 10.1101/2025.03.12.642788. bioRxiv. 2025. Update in: Genetics. 2025 Jul 31:iyaf147. doi: 10.1093/genetics/iyaf147. PMID: 40161657 Free PMC article. Updated. Preprint.
-
In renal proximal tubular epithelial cells of the hibernator Syrian hamster, anoxia-reoxygenation-induced reactive oxygen species bursts do not trigger a DNA damage response and cellular senescence.J Comp Physiol B. 2025 Feb;195(1):91-101. doi: 10.1007/s00360-025-01604-5. Epub 2025 Feb 5. J Comp Physiol B. 2025. PMID: 39907746 Free PMC article.
-
The Role of Endothelial Cell Mitophagy in Age-Related Cardiovascular Diseases.Aging Dis. 2024 Jul 26;16(4):2151-2176. doi: 10.14336/AD.2024.0788. Aging Dis. 2024. PMID: 39122456 Free PMC article. Review.
-
Amyloid β instigates cardiac neurotrophic signaling impairment, driving Alzheimer's associated heart disease.bioRxiv [Preprint]. 2025 Jun 4:2023.07.11.548558. doi: 10.1101/2023.07.11.548558. bioRxiv. 2025. PMID: 37502936 Free PMC article. Preprint.
References
-
- Hauptmann S., Scherping I., Dröse S., Brandt U., Schulz K.L., Jendrach M., Leuner K., Eckert A., Müller W.E. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging. 2009;30:1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
