

Epigenetic Regulation of Neuroinflammation in Alzheimer's Disease
- PMID: 38201283
- PMCID: PMC10778497
- DOI: 10.3390/cells13010079
Epigenetic Regulation of Neuroinflammation in Alzheimer's Disease
Abstract
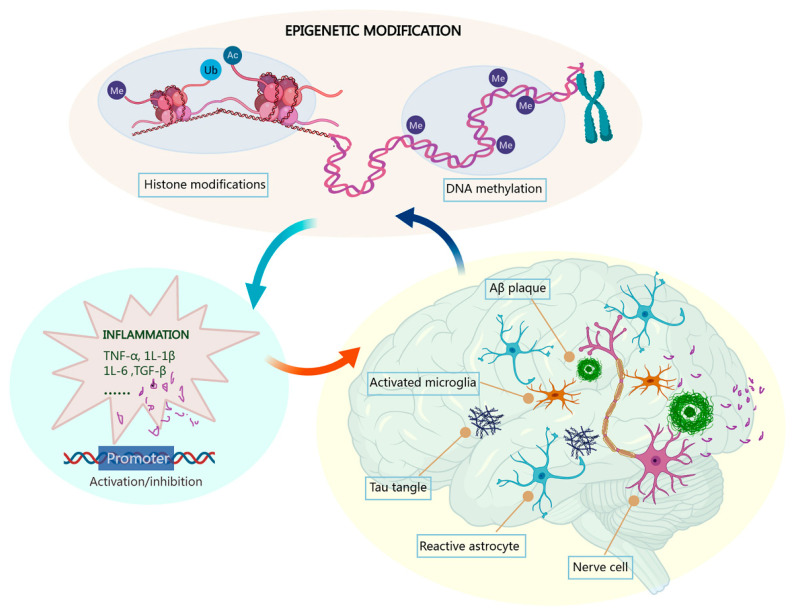
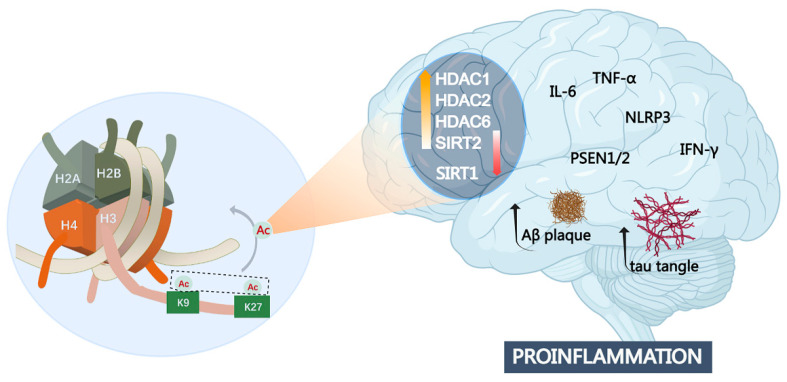
Alzheimer's disease (AD) is a chronic and progressive neurodegenerative disease and clinically manifests with cognitive decline and behavioral disabilities. Over the past years, mounting studies have demonstrated that the inflammatory response plays a key role in the onset and development of AD, and neuroinflammation has been proposed as the third major pathological driving factor of AD, ranking after the two well-known core pathologies, amyloid β (Aβ) deposits and neurofibrillary tangles (NFTs). Epigenetic mechanisms, referring to heritable changes in gene expression independent of DNA sequence alterations, are crucial regulators of neuroinflammation which have emerged as potential therapeutic targets for AD. Upon regulation of transcriptional repression or activation, epigenetic modification profiles are closely involved in inflammatory gene expression and signaling pathways of neuronal differentiation and cognitive function in central nervous system disorders. In this review, we summarize the current knowledge about epigenetic control mechanisms with a focus on DNA and histone modifications involved in the regulation of inflammatory genes and signaling pathways in AD, and the inhibitors under clinical assessment are also discussed.
Keywords: Alzheimer’s disease; DNA methylation; epigenetics; histone modification; inflammation; microglia.
Conflict of interest statement
The authors declare no conflicts of interest, financial or otherwise.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
