

KLC1-ROS1 Fusion Exerts Oncogenic Properties of Glioma Cells via Specific Activation of JAK-STAT Pathway
- PMID: 38201436
- PMCID: PMC10778328
- DOI: 10.3390/cancers16010009
KLC1-ROS1 Fusion Exerts Oncogenic Properties of Glioma Cells via Specific Activation of JAK-STAT Pathway
Abstract
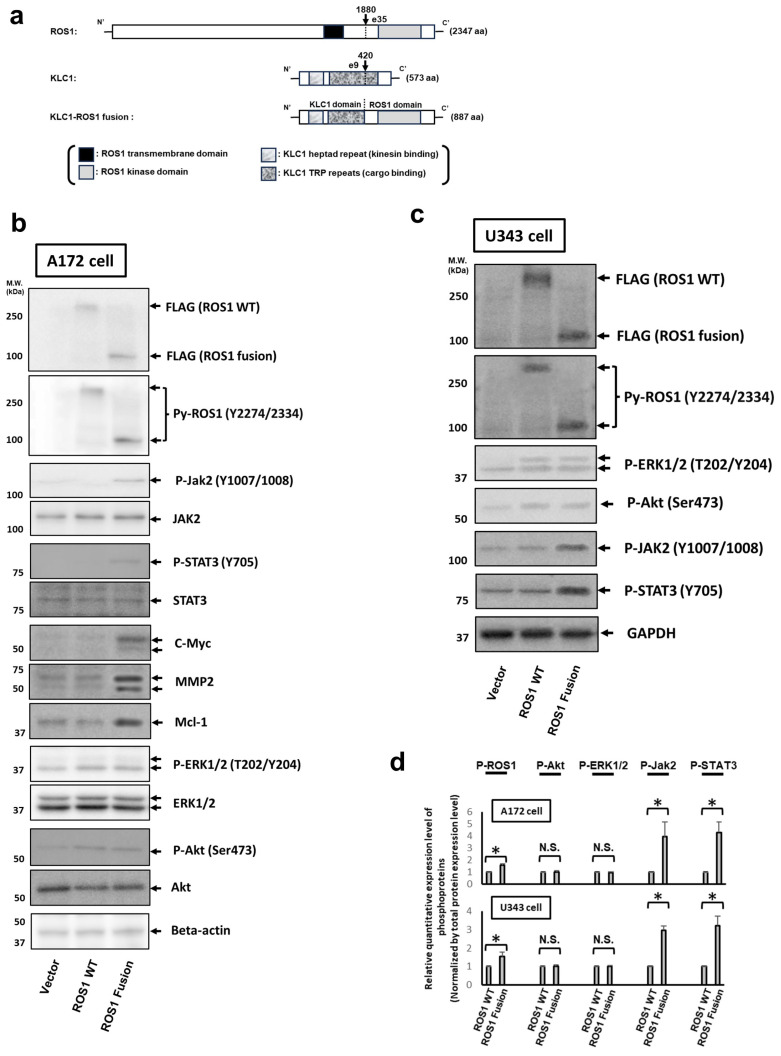
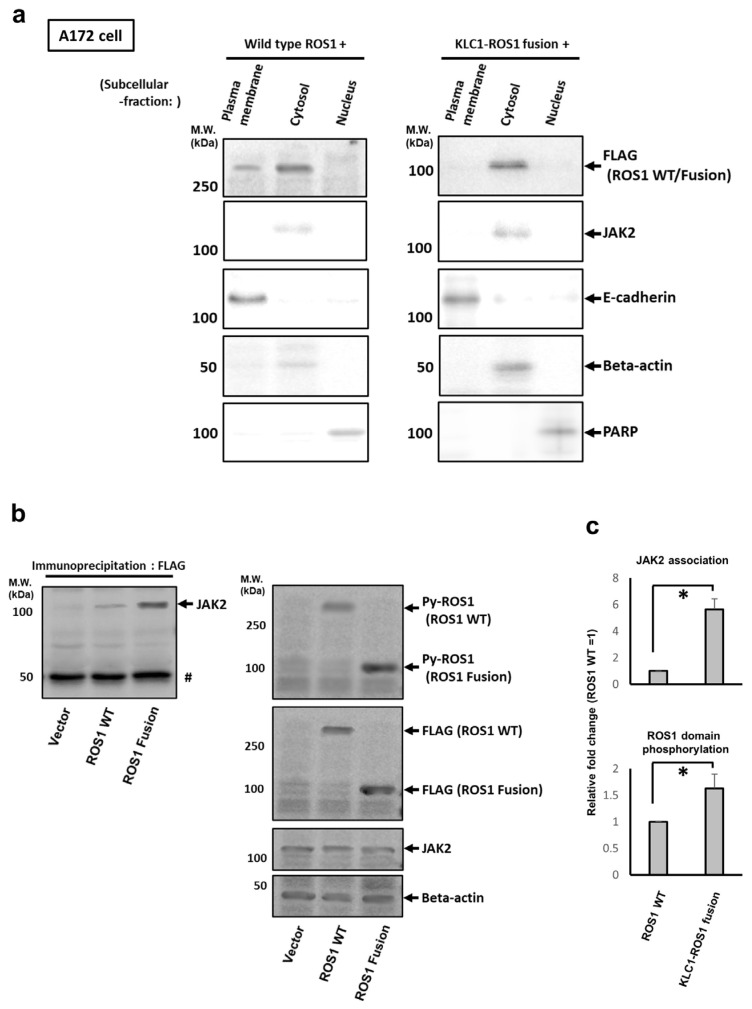
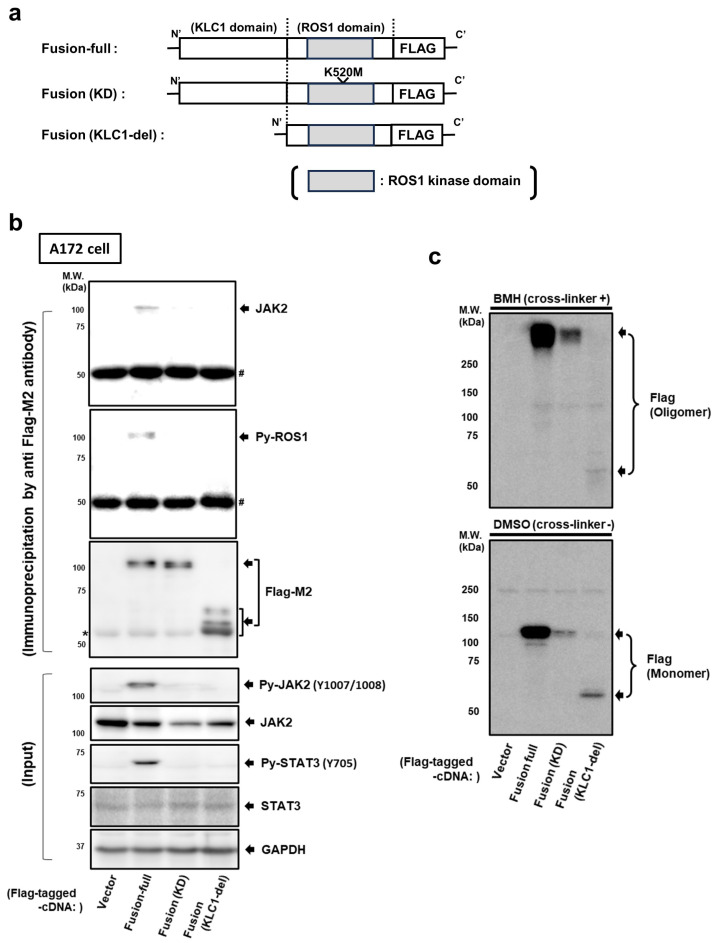
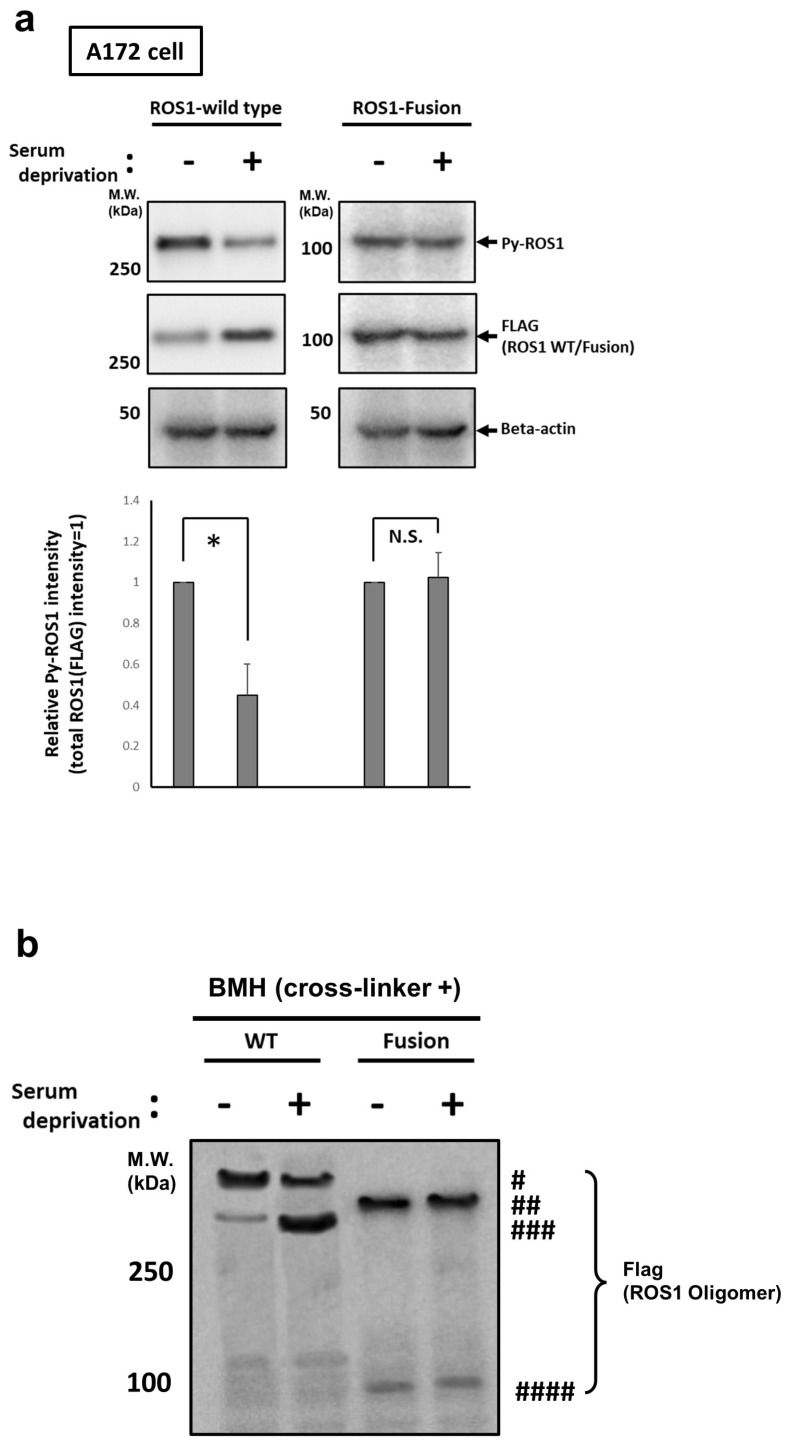
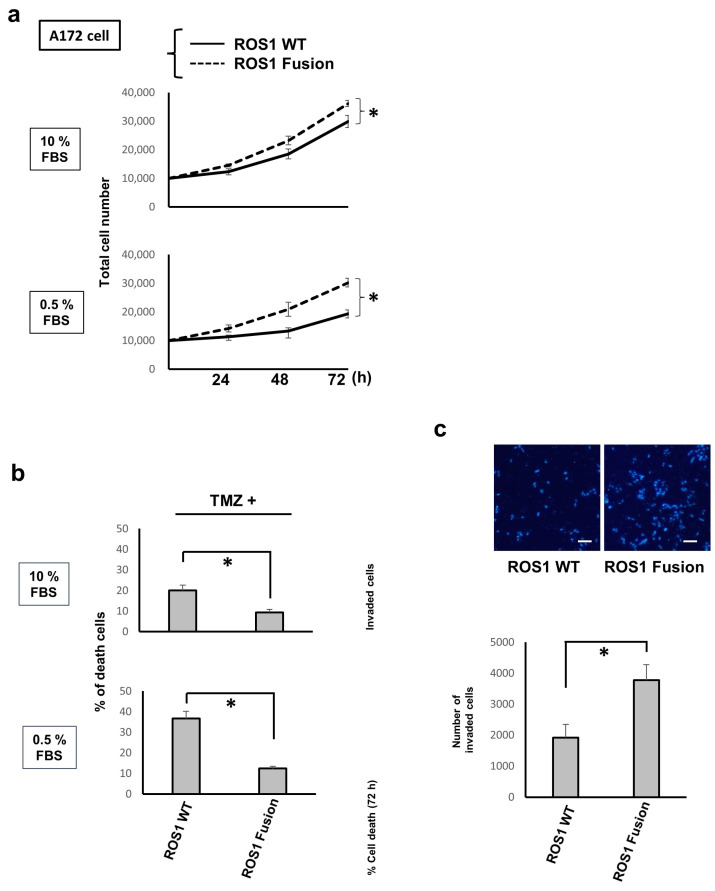
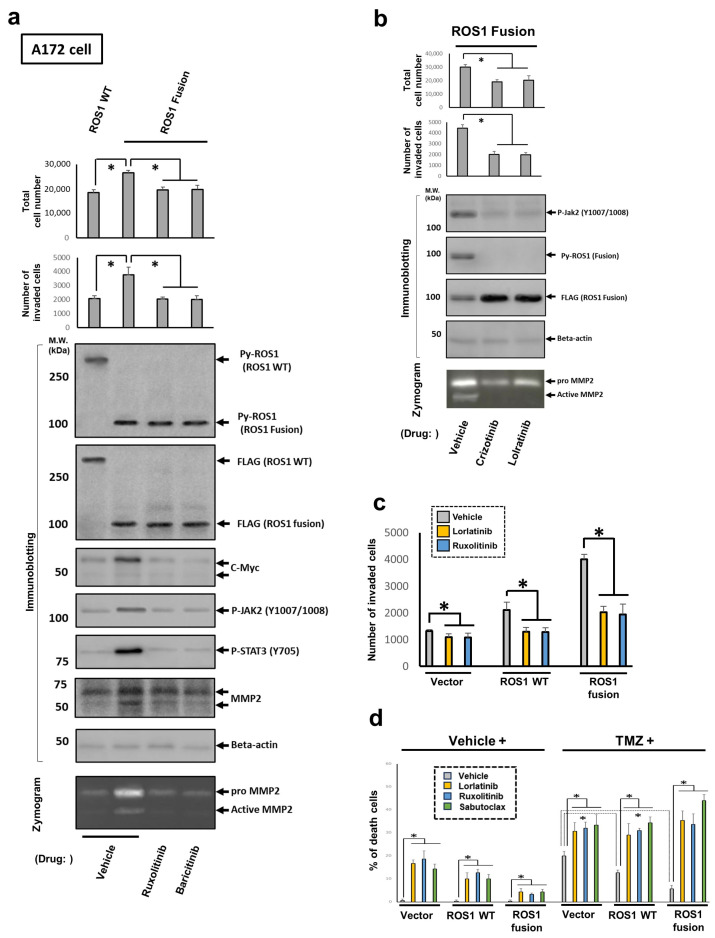
Here, we investigated the detailed molecular oncogenic mechanisms of a novel receptor tyrosine kinase (RTK) fusion, KLC1-ROS1, with an adapter molecule, KLC1, and an RTK, ROS1, discovered in pediatric glioma, and we explored a novel therapeutic target for glioma that possesses oncogenic RTK fusion. When wild-type ROS1 and KLC1-ROS1 fusions were stably expressed in the human glioma cell lines A172 and U343MG, immunoblotting revealed that KLC1-ROS1 fusion specifically activated the JAK2-STAT3 pathway, a major RTK downstream signaling pathway, when compared with wild-type ROS1. Immunoprecipitation of the fractionated cell lysates revealed a more abundant association of the KLC1-ROS1 fusion with JAK2 than that observed for wild-type ROS1 in the cytosolic fraction. A mutagenesis study of the KLC1-ROS1 fusion protein demonstrated the fundamental roles of both the KLC1 and ROS1 domains in the constitutive activation of KLC1-ROS1 fusion. Additionally, in vitro assays demonstrated that KLC1-ROS1 fusion upregulated cell proliferation, invasion, and chemoresistance when compared to wild-type ROS1. Combination treatment with the chemotherapeutic agent temozolomide and an inhibitor of ROS1, JAK2, or a downstream target of STAT3, demonstrated antitumor effects against KLC1-ROS1 fusion-expressing glioma cells. Our results demonstrate that KLC1-ROS1 fusion exerts oncogenic activity through serum-independent constitutive activation, resulting in specific activation of the JAK-STAT pathway. Our data suggested that molecules other than RTKs may serve as novel therapeutic targets for RTK fusion in gliomas.
Keywords: JAK-STAT pathway; KLC1-ROS1 fusion; glioma; oncogene.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Identification of a novel KLC1-ROS1 fusion in a case of pediatric low-grade localized glioma.Brain Tumor Pathol. 2019 Jan;36(1):14-19. doi: 10.1007/s10014-018-0330-3. Epub 2018 Oct 22. Brain Tumor Pathol. 2019. PMID: 30350109 Review.
-
Novel TPR::ROS1 Fusion Gene Activates MAPK, PI3K and JAK/STAT Signaling in an Infant-type Pediatric Glioma.Cancer Genomics Proteomics. 2022 Nov-Dec;19(6):711-726. doi: 10.21873/cgp.20354. Cancer Genomics Proteomics. 2022. PMID: 36316040 Free PMC article.
-
Identification of a Novel MAN1A1-ROS1 Fusion Gene Through mRNA-based Screening for Tyrosine Kinase Gene Aberrations in a Patient with Leiomyosarcoma.Clin Orthop Relat Res. 2021 Apr 1;479(4):838-852. doi: 10.1097/CORR.0000000000001548. Clin Orthop Relat Res. 2021. PMID: 33196586 Free PMC article.
-
Novel insight into mechanisms of ROS1 catalytic activation via loss of the extracellular domain.Sci Rep. 2024 Sep 27;14(1):22191. doi: 10.1038/s41598-024-71687-7. Sci Rep. 2024. PMID: 39333184 Free PMC article.
-
ROS1 protein-tyrosine kinase inhibitors in the treatment of ROS1 fusion protein-driven non-small cell lung cancers.Pharmacol Res. 2017 Jul;121:202-212. doi: 10.1016/j.phrs.2017.04.022. Epub 2017 Apr 30. Pharmacol Res. 2017. PMID: 28465216 Review.
Cited by
-
Quantitative proteomics analysis reveals possible anticancer mechanisms of 5'-deoxy-5'-methylthioadenosine in cholangiocarcinoma cells.PLoS One. 2024 Jun 26;19(6):e0306060. doi: 10.1371/journal.pone.0306060. eCollection 2024. PLoS One. 2024. PMID: 38923999 Free PMC article.
-
Comprehensive analysis of the prognostic and immunological signature of TNFAIP8 family genes in human glioma.Sci Rep. 2024 Aug 2;14(1):17875. doi: 10.1038/s41598-024-68784-y. Sci Rep. 2024. PMID: 39090168 Free PMC article.
-
Decoding the Role of Kinesin Superfamily Proteins in Glioma Progression.J Mol Neurosci. 2025 Jan 23;75(1):10. doi: 10.1007/s12031-025-02308-9. J Mol Neurosci. 2025. PMID: 39847238 Review.
-
Oncogenic gene fusions in cancer: from biology to therapy.Signal Transduct Target Ther. 2025 Apr 14;10(1):111. doi: 10.1038/s41392-025-02161-7. Signal Transduct Target Ther. 2025. PMID: 40223139 Free PMC article. Review.
-
The supramolecular polymer-related signature predicts prognosis and indicates immune microenvironment infiltration in gastric cancer.Clinics (Sao Paulo). 2025 Apr 13;80:100641. doi: 10.1016/j.clinsp.2025.100641. eCollection 2025. Clinics (Sao Paulo). 2025. PMID: 40228435 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Miscellaneous
