

Phogrin Regulates High-Fat Diet-Induced Compensatory Pancreatic β-Cell Growth by Switching Binding Partners
- PMID: 38201998
- PMCID: PMC10780347
- DOI: 10.3390/nu16010169
Phogrin Regulates High-Fat Diet-Induced Compensatory Pancreatic β-Cell Growth by Switching Binding Partners
Abstract
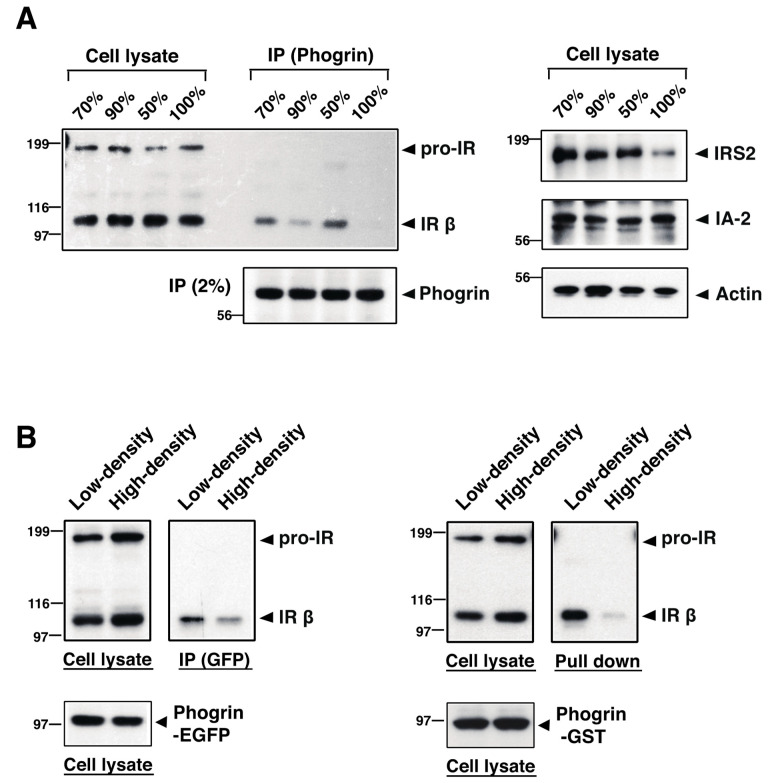
The receptor protein tyrosine phosphatase phogrin primarily localizes to hormone secretory granules in neuroendocrine cells. Concurrent with glucose-stimulated insulin secretion, phogrin translocates to pancreatic β-cell plasma membranes, where it interacts with insulin receptors (IRs) to stabilize insulin receptor substrate 2 (IRS2) that, in turn, contributes to glucose-responsive β-cell growth. Pancreatic β-cell development was not altered in β-cell-specific, phogrin-deficient mice, but the thymidine incorporation rate decreased in phogrin-deficient islets with a moderate reduction in IRS2 protein expression. In this study, we analyzed the β-cell response to high-fat diet stress and found that the compensatory expansion in β-cell mass was significantly suppressed in phogrin-deficient mice. Phogrin-IR interactions occurred only in high-fat diet murine islets and proliferating β-cell lines, whereas they were inhibited by the intercellular binding of surface phogrin under confluent cell culture conditions. Thus, phogrin could regulate glucose-stimulated compensatory β-cell growth by changing its binding partner from another β-cell phogrin to IR in the same β-cells.
Keywords: high-fat diet; insulin receptor substrate; insulin signaling; islet antigen; pancreatic β-cell mass; secretory granules.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures





Similar articles
-
The pseudophosphatase phogrin enables glucose-stimulated insulin signaling in pancreatic β cells.J Biol Chem. 2018 Apr 20;293(16):5920-5933. doi: 10.1074/jbc.RA117.000301. Epub 2018 Feb 26. J Biol Chem. 2018. PMID: 29483197 Free PMC article.
-
Gene silencing of phogrin unveils its essential role in glucose-responsive pancreatic beta-cell growth.Diabetes. 2009 Mar;58(3):682-92. doi: 10.2337/db08-0970. Epub 2008 Dec 10. Diabetes. 2009. PMID: 19073770 Free PMC article.
-
Different regulated expression of the tyrosine phosphatase-like proteins IA-2 and phogrin by glucose and insulin in pancreatic islets: relationship to development of insulin secretory responses in early life.Diabetes. 2002 Oct;51(10):2982-8. doi: 10.2337/diabetes.51.10.2982. Diabetes. 2002. PMID: 12351437
-
Insulin granule morphology and crinosome formation in mice lacking the pancreatic β cell-specific phogrin (PTPRN2) gene.Histochem Cell Biol. 2024 Mar;161(3):223-238. doi: 10.1007/s00418-023-02256-8. Epub 2023 Dec 27. Histochem Cell Biol. 2024. PMID: 38150052
-
Secretagogue-dependent phosphorylation of phogrin, an insulin granule membrane protein tyrosine phosphatase homologue.Biochem J. 1999 Aug 1;341 ( Pt 3)(Pt 3):563-9. Biochem J. 1999. PMID: 10417318 Free PMC article.
References
-
- Lu J., Li Q., Xie H., Chen Z.J., Borovitskaya A.E., Maclaren N.K., Notkins A.L., Lan M.S. Identification of a second transmembrane protein tyrosine phosphatase, IA-2beta, as an autoantigen in insulin-dependent diabetes mellitus: Precursor of the 37-kDa tryptic fragment. Proc. Natl. Acad. Sci. USA. 1996;93:2307–2311. doi: 10.1073/pnas.93.6.2307. - DOI - PMC - PubMed
-
- Cui L., Yu W., DeAizpurua H.J., Schmidli R.S., Pallen C.J. Cloning and characterization of islet cell antigen-related protein-tyrosine phosphatase (PTP), a novel receptor-like PTP and autoantigen in insulin-dependent diabetes. J. Biol. Chem. 1996;271:24817–24823. doi: 10.1074/jbc.271.40.24817. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
