

"NO" Time in Fear Response: Possible Implication of Nitric-Oxide-Related Mechanisms in PTSD
- PMID: 38202672
- PMCID: PMC10779493
- DOI: 10.3390/molecules29010089
"NO" Time in Fear Response: Possible Implication of Nitric-Oxide-Related Mechanisms in PTSD
Abstract
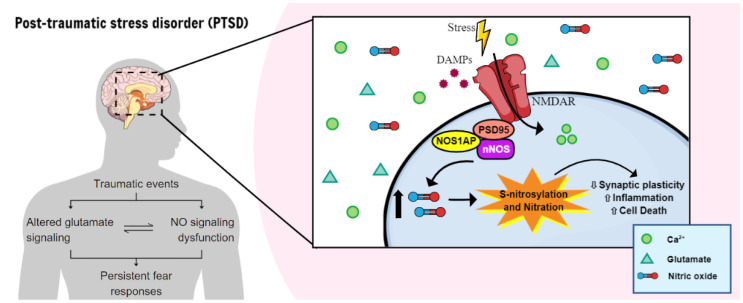
Post-traumatic stress disorder (PTSD) is a psychiatric condition characterized by persistent fear responses and altered neurotransmitter functioning due to traumatic experiences. Stress predominantly affects glutamate, a neurotransmitter crucial for synaptic plasticity and memory formation. Activation of the N-Methyl-D-Aspartate glutamate receptors (NMDAR) can trigger the formation of a complex comprising postsynaptic density protein-95 (PSD95), the neuronal nitric oxide synthase (nNOS), and its adaptor protein (NOS1AP). This complex is pivotal in activating nNOS and nitric oxide (NO) production, which, in turn, activates downstream pathways that modulate neuronal signaling, including synaptic plasticity/transmission, inflammation, and cell death. The involvement of nNOS and NOS1AP in the susceptibility of PTSD and its comorbidities has been widely shown. Therefore, understanding the interplay between stress, fear, and NO is essential for comprehending the maintenance and progression of PTSD, since NO is involved in fear acquisition and extinction processes. Moreover, NO induces post-translational modifications (PTMs), including S-nitrosylation and nitration, which alter protein function and structure for intracellular signaling. Although evidence suggests that NO influences synaptic plasticity and memory processing, the specific role of PTMs in the pathophysiology of PTSD remains unclear. This review highlights pathways modulated by NO that could be relevant to stress and PTSD.
Keywords: memory; nNOS; post-translational modifications.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Figures



Similar articles
-
Targeting neuronal nitric oxide synthase and the nitrergic system in post-traumatic stress disorder.Psychopharmacology (Berl). 2022 Oct;239(10):3057-3082. doi: 10.1007/s00213-022-06212-7. Epub 2022 Aug 27. Psychopharmacology (Berl). 2022. PMID: 36029333 Review.
-
PSD95 and nNOS interaction as a novel molecular target to modulate conditioned fear: relevance to PTSD.Transl Psychiatry. 2018 Aug 14;8(1):155. doi: 10.1038/s41398-018-0208-5. Transl Psychiatry. 2018. PMID: 30108200 Free PMC article.
-
Coupling between neuronal nitric oxide synthase and glutamate receptor 6-mediated c-Jun N-terminal kinase signaling pathway via S-nitrosylation contributes to ischemia neuronal death.Neuroscience. 2008 Sep 9;155(4):1120-32. doi: 10.1016/j.neuroscience.2008.03.061. Epub 2008 Apr 4. Neuroscience. 2008. PMID: 18676085
-
Pharmacological rewriting of fear memories: A beacon for post-traumatic stress disorder.Eur J Pharmacol. 2020 Mar 5;870:172824. doi: 10.1016/j.ejphar.2019.172824. Epub 2019 Nov 25. Eur J Pharmacol. 2020. PMID: 31778672 Review.
-
Disruption of nNOS-NOS1AP protein-protein interactions suppresses neuropathic pain in mice.Pain. 2018 May;159(5):849-863. doi: 10.1097/j.pain.0000000000001152. Pain. 2018. PMID: 29319606 Free PMC article.
Cited by
-
The molecular mechanism of nitric oxide in memory consolidation and its role in the pathogenesis of memory-related disorders.Neurogenetics. 2025 Jan 24;26(1):22. doi: 10.1007/s10048-025-00803-0. Neurogenetics. 2025. PMID: 39853459 Free PMC article. Review.
-
Function and application of brain‑derived neurotrophic factor precursors (Review).Int J Mol Med. 2025 Jul;56(1):105. doi: 10.3892/ijmm.2025.5546. Epub 2025 May 9. Int J Mol Med. 2025. PMID: 40341415 Free PMC article. Review.
References
-
- Bremner J.D., Staib L.H., Kaloupek D., Southwick S.M., Soufer R., Charney D.S. Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: A positron emission tomography study. Biol. Psychiatry. 1999;45:806–816. doi: 10.1016/S0006-3223(98)00297-2. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
