

Bidirectional Allosteric Coupling between PIP2 Binding and the Pore of the Oncochannel TRPV6
- PMID: 38203789
- PMCID: PMC10779433
- DOI: 10.3390/ijms25010618
Bidirectional Allosteric Coupling between PIP2 Binding and the Pore of the Oncochannel TRPV6
Abstract
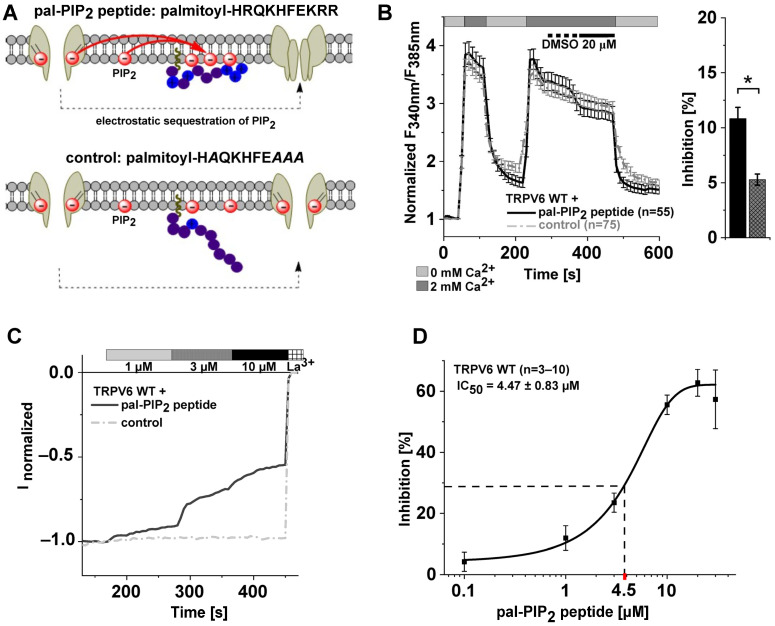
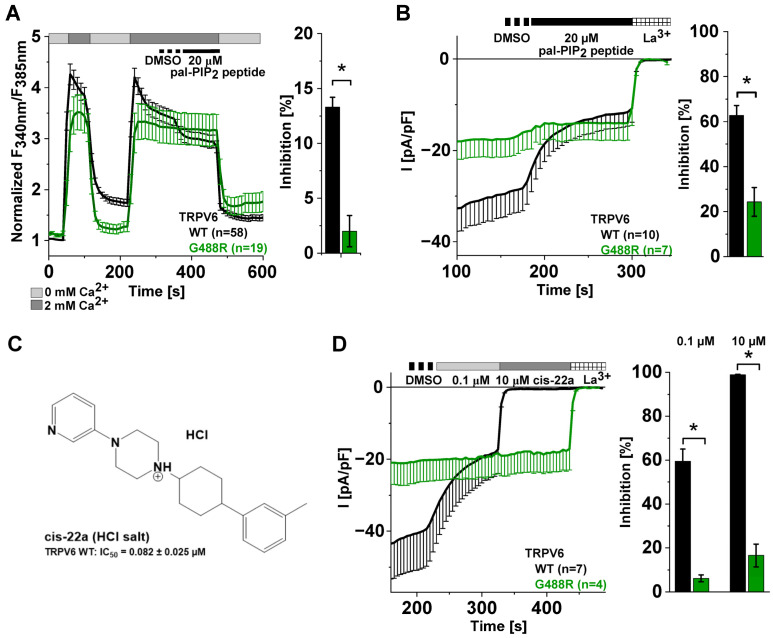
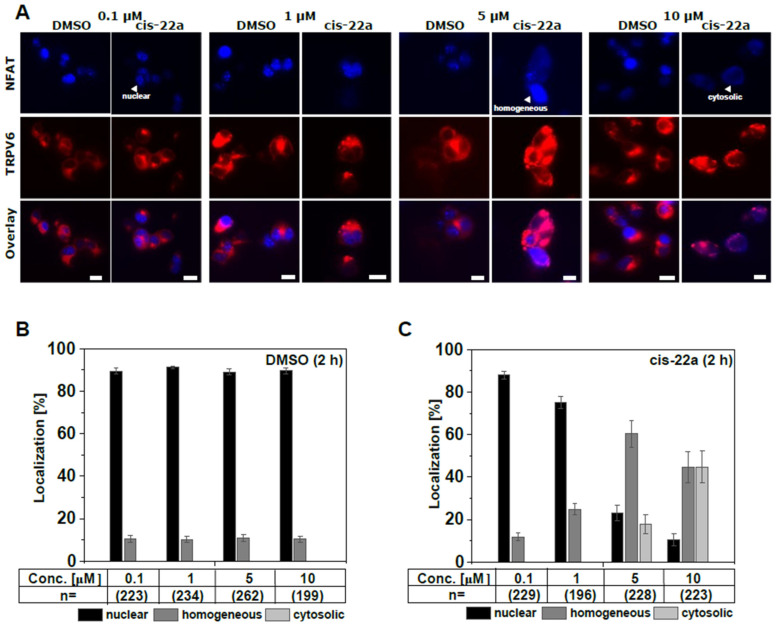
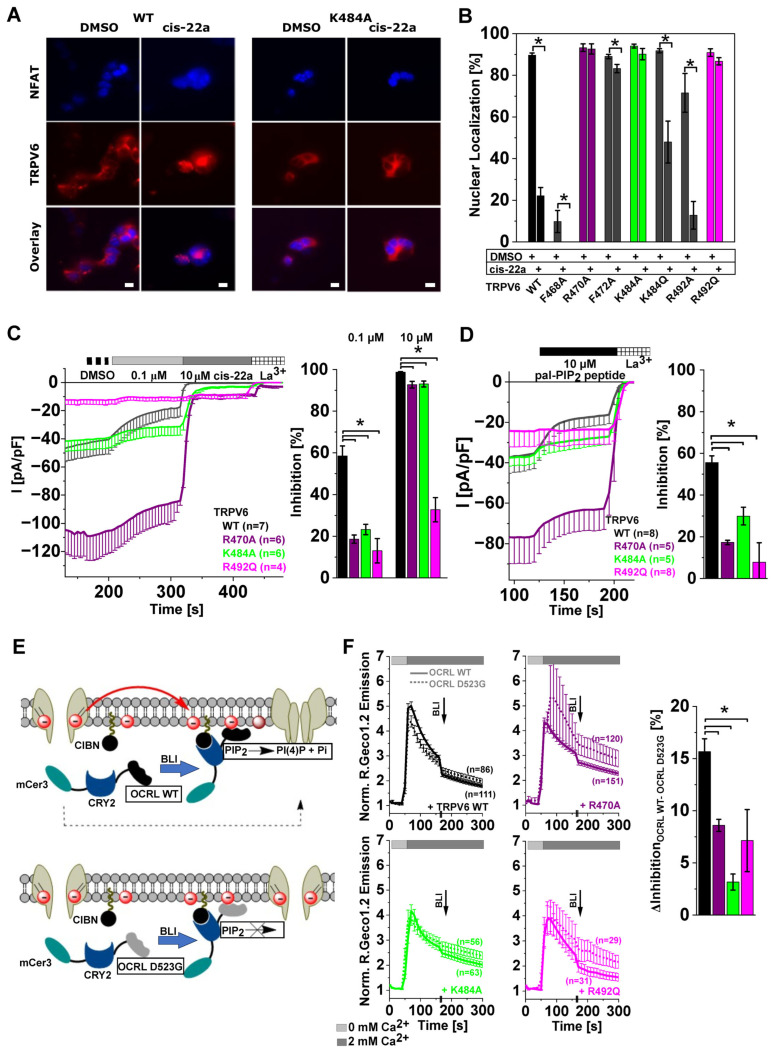
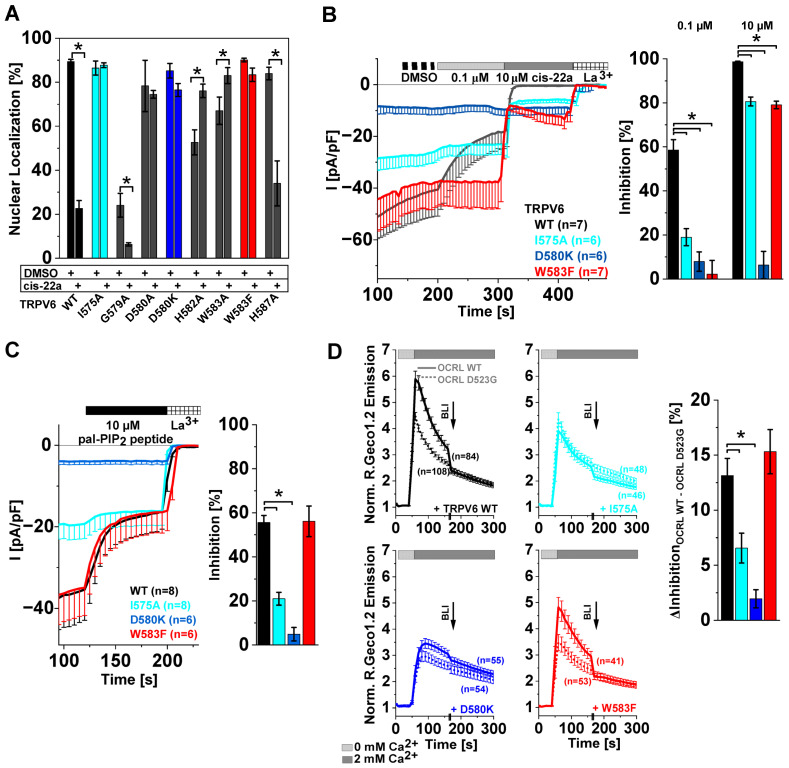
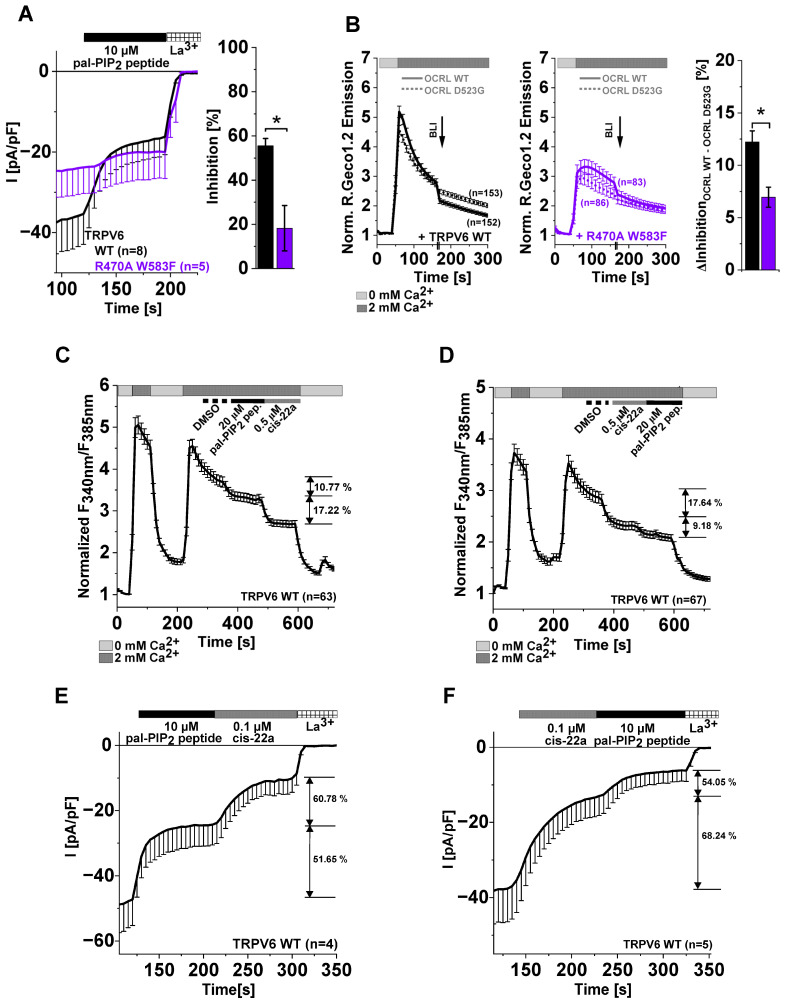
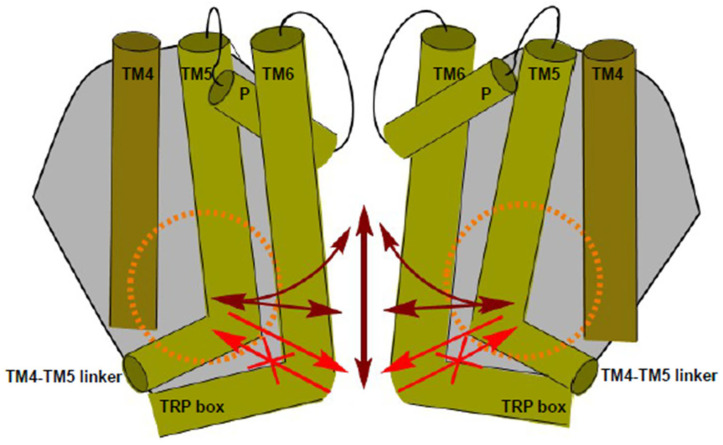
The epithelial ion channel TRPV6 plays a pivotal role in calcium homeostasis. Channel function is intricately regulated at different stages, involving the lipid phosphatidylinositol-4,5-bisphosphate (PIP2). Given that dysregulation of TRPV6 is associated with various diseases, including different types of cancer, there is a compelling need for its pharmacological targeting. Structural studies provide insights on how TRPV6 is affected by different inhibitors, with some binding to sites else occupied by lipids. These include the small molecule cis-22a, which, however, also binds to and thereby blocks the pore. By combining calcium imaging, electrophysiology and optogenetics, we identified residues within the pore and the lipid binding site that are relevant for regulation by cis-22a and PIP2 in a bidirectional manner. Yet, mutation of the cytosolic pore exit reduced inhibition by cis-22a but preserved sensitivity to PIP2 depletion. Our data underscore allosteric communication between the lipid binding site and the pore and vice versa for most sites along the pore.
Keywords: PIP2; TRPV6; calcium; cis-22a; inhibition; lipid.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
TRPV6 Regulation by Cis-22a and Cholesterol.Biomolecules. 2022 Jun 8;12(6):804. doi: 10.3390/biom12060804. Biomolecules. 2022. PMID: 35740929 Free PMC article.
-
Molecular pathway and structural mechanism of human oncochannel TRPV6 inhibition by the phytocannabinoid tetrahydrocannabivarin.Nat Commun. 2023 Aug 2;14(1):4630. doi: 10.1038/s41467-023-40362-2. Nat Commun. 2023. PMID: 37532722 Free PMC article.
-
Structure and function of the calcium-selective TRP channel TRPV6.J Physiol. 2021 May;599(10):2673-2697. doi: 10.1113/JP279024. Epub 2020 Mar 13. J Physiol. 2021. PMID: 32073143 Free PMC article. Review.
-
Inactivation-mimicking block of the epithelial calcium channel TRPV6.Sci Adv. 2020 Nov 27;6(48):eabe1508. doi: 10.1126/sciadv.abe1508. Print 2020 Nov. Sci Adv. 2020. PMID: 33246965 Free PMC article.
-
Molecular pharmacology of the onco-TRP channel TRPV6.Channels (Austin). 2023 Dec;17(1):2266669. doi: 10.1080/19336950.2023.2266669. Epub 2023 Oct 15. Channels (Austin). 2023. PMID: 37838981 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
