

Extracellular Matrix Protein-1 as a Mediator of Inflammation-Induced Fibrosis After Myocardial Infarction
- PMID: 38205347
- PMCID: PMC10774582
- DOI: 10.1016/j.jacbts.2023.05.010
Extracellular Matrix Protein-1 as a Mediator of Inflammation-Induced Fibrosis After Myocardial Infarction
Abstract
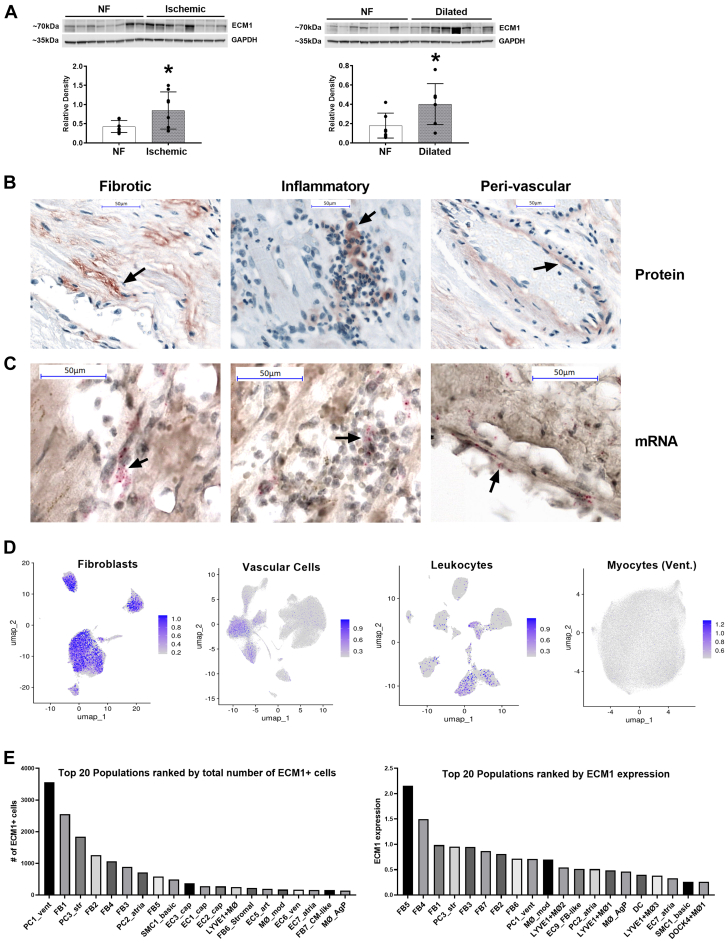
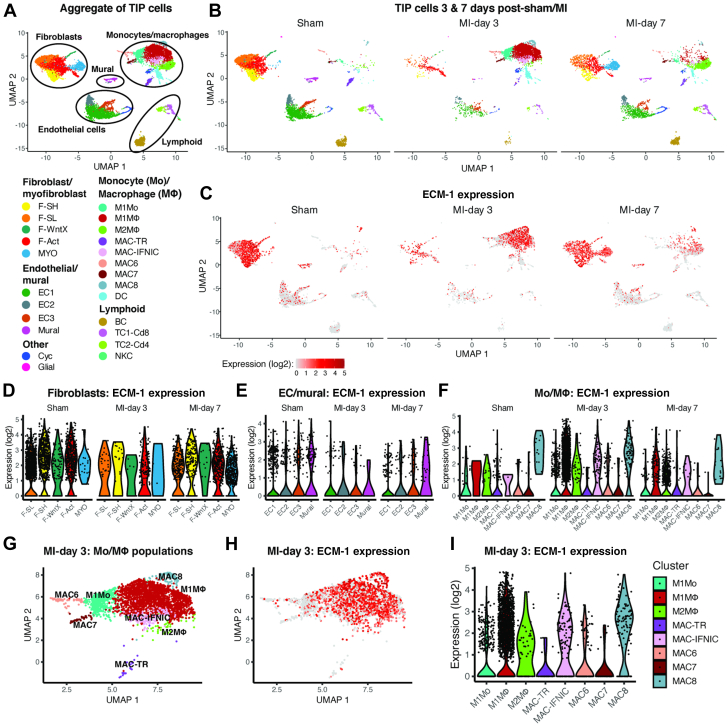
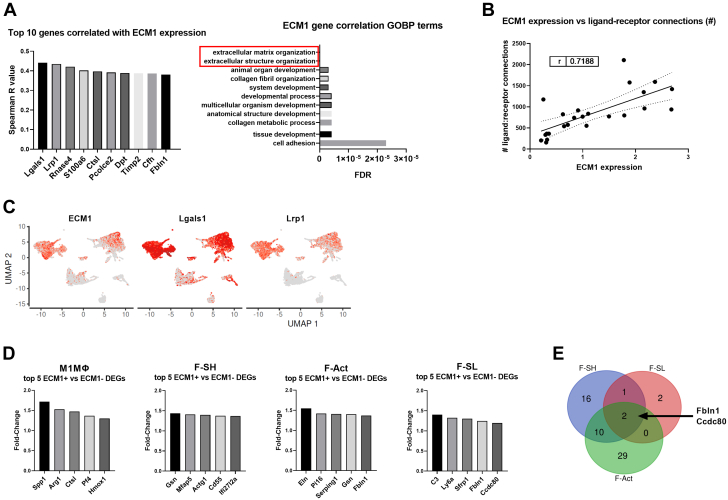
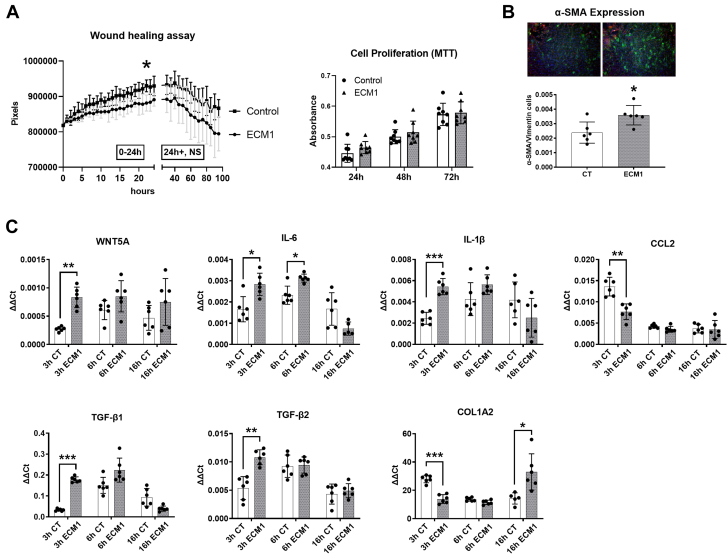
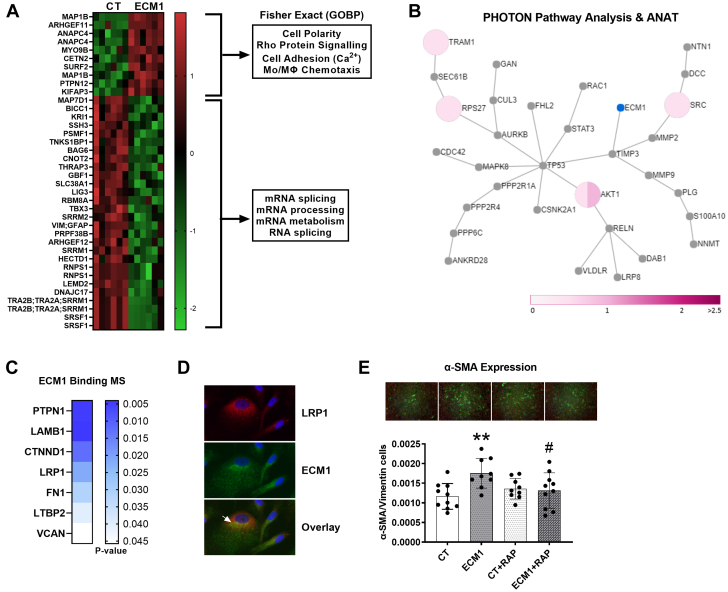
Irreversible fibrosis is a hallmark of myocardial infarction (MI) and heart failure. Extracellular matrix protein-1 (ECM-1) is up-regulated in these hearts, localized to fibrotic, inflammatory, and perivascular areas. ECM-1 originates predominantly from fibroblasts, macrophages, and pericytes/vascular cells in uninjured human and mouse hearts, and from M1 and M2 macrophages and myofibroblasts after MI. ECM-1 stimulates fibroblast-to-myofibroblast transition, up-regulates key fibrotic and inflammatory pathways, and inhibits cardiac fibroblast migration. ECM-1 binds HuCFb cell surface receptor LRP1, and LRP1 inhibition blocks ECM-1 from stimulating fibroblast-to-myofibroblast transition, confirming a novel ECM-1-LRP1 fibrotic signaling axis. ECM-1 may represent a novel mechanism facilitating inflammation-fibrosis crosstalk.
Keywords: extracellular matrix; fibroblasts; fibrosis; heart; inflammation; myocardial infarction.
© 2023 The Authors.
Conflict of interest statement
This work was supported by the Austrian Society of Cardiology, ERA-CVD, and Austrian Science Fund AIR-MI consortium (I 4168-B to Dr Rainer); John Hunter Charitable Trust (G1800510 to Dr Boyle); Hunter Medical Research Institute and Emlyn and Jennie Thomas Postgraduate Medical Research Scholarship (G2100164 and G1800696 to Dr Boyle); Australian Commonwealth funded Research Training Program stipend (to Drs Hardy and Mabotuwana); Medical University of Graz Doctoral School of Translational Molecular and Cellular Biosciences [to Drs Hardy and Mabotuwana] and Doctoral School of Molecular Medicine [to Dr Rech]; Austrian Society of Cardiology (to Dr Hardy); National Health and Medical Research Council (NHMRC) (2000615 and 1074386 to Dr Harvey, 1079187 and 1175134 to Dr Hansbro, and 1156898 and 20000483 to Dr Starkey); NHMRC Senior Principal Research Fellowship (1118576 to Dr Harvey); Stem Cells Australia (SR110001002 to Dr Harvey); the Victor Chang Cardiac Research Institute, Australia [to Dr Harvey]; the University of Technology Sydney, Australia [to Dr Hansbro]; Monash University, Australia (to Dr Starkey); Australian Research Council (DE170100226 to Dr Starkey); Foundation Leducq (15CVD03 and 13CVD01 to Dr Harvey); Austrian Science fund (KLI645, W1226, and F73 to Dr Birner-Gruenberger); Interdisciplinary Centre for Clinical Research, University Hospital Würzburg (E-353 to Dr Cochain, E-354 to Dr Campos Ramos); the German Research Foundation [471705758 and 458539578 to Dr Cochain, DFG SFB1525 project no. 453989101 to Drs Cochain and Campos Ramos, and 411619907 to Dr Campos Ramos]; and the European Research Area Network-Cardiovascular Diseases/German Federal Ministry of Education and Research (01KL1902 to Dr Campos Ramos). All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.PerspectivesCOMPETENCY IN MEDICAL KNOWLEDGE: Fibrosis is a key factor in heart disease that impedes cardiac function and prognosis. Yet, no therapies specifically target profibrotic signaling in the heart, and once established, fibrotic remodeling is largely irreversible. Inflammation-fibrosis crosstalk, whereby ECM remodeling and fibrotic tissue deposition is tightly connected to inflammation and vice versa, is now thought to be critical in orchestrating wound healing and cardiac scarring. It is suggested that this is why unidirectional therapies targeting fibrosis or inflammation alone have failed to substantially reduce the mortality associated with cardiac diseases. Here, we represent ECM-1 as a potential novel mediator of inflammation-induced fibrotic signaling in the heart and confirm its regulation in human heart failure. TRANSLATIONAL OUTLOOK: ECM-1 may serve as an attractive future treatment target to prevent excessive and detrimental fibrosis. Because fibrosis and scarring is a universal response to injury in many organs, this has potential implications beyond heart disease.
Figures
References
-
- Forte E., Furtado M.B., Rosenthal N. The interstitium in cardiac repair: role of the immune–stromal cell interplay. Nat Rev Cardiol. 2018;15:601–616. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
