

A new genomic framework to categorize pediatric acute myeloid leukemia
- PMID: 38212634
- PMCID: PMC10864188
- DOI: 10.1038/s41588-023-01640-3
A new genomic framework to categorize pediatric acute myeloid leukemia
Abstract
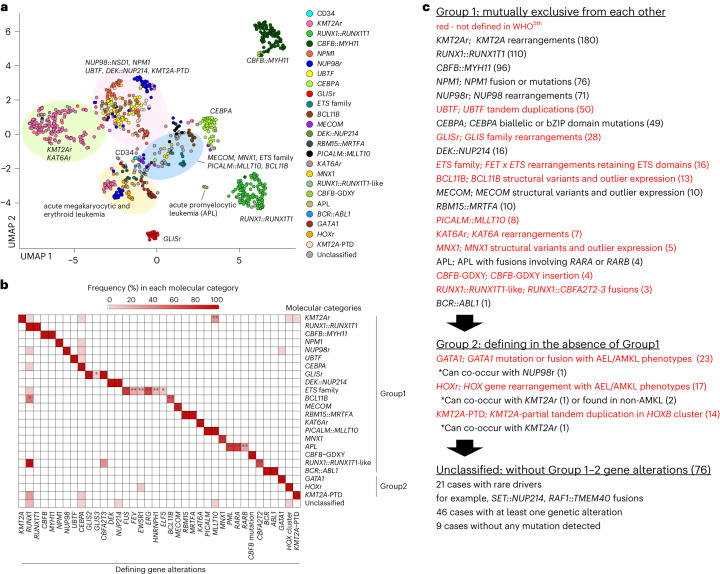
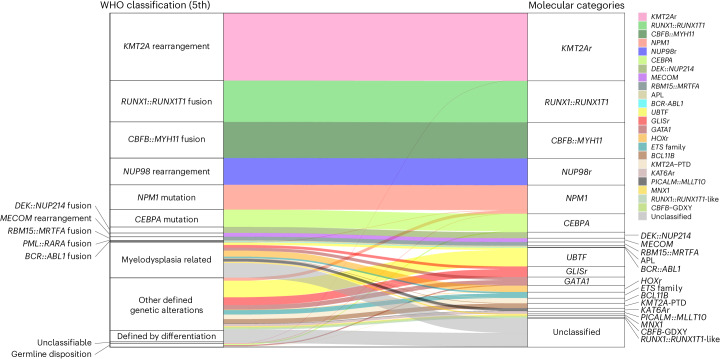
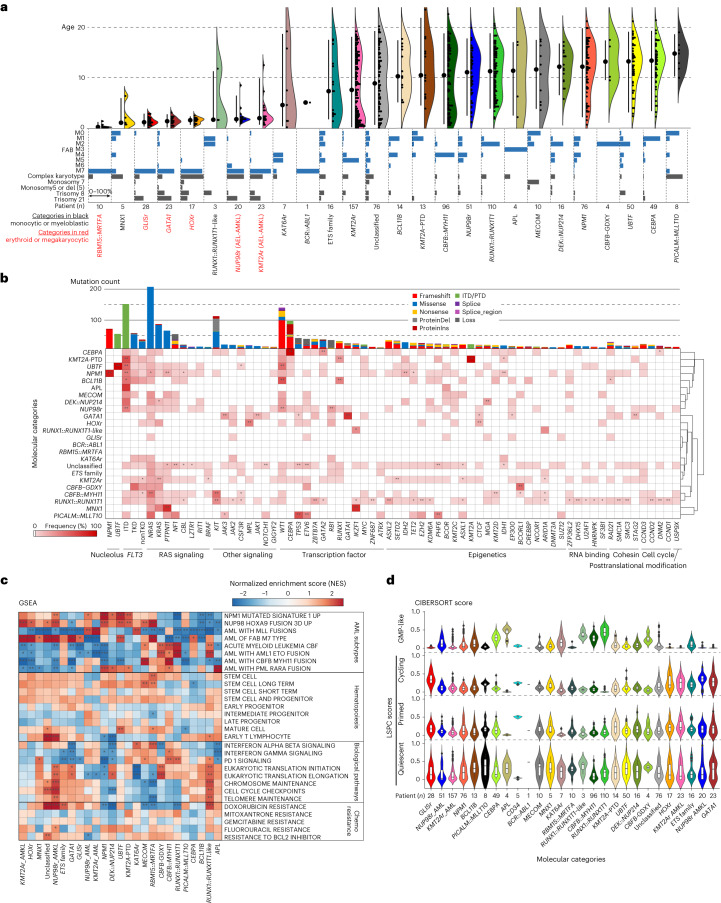
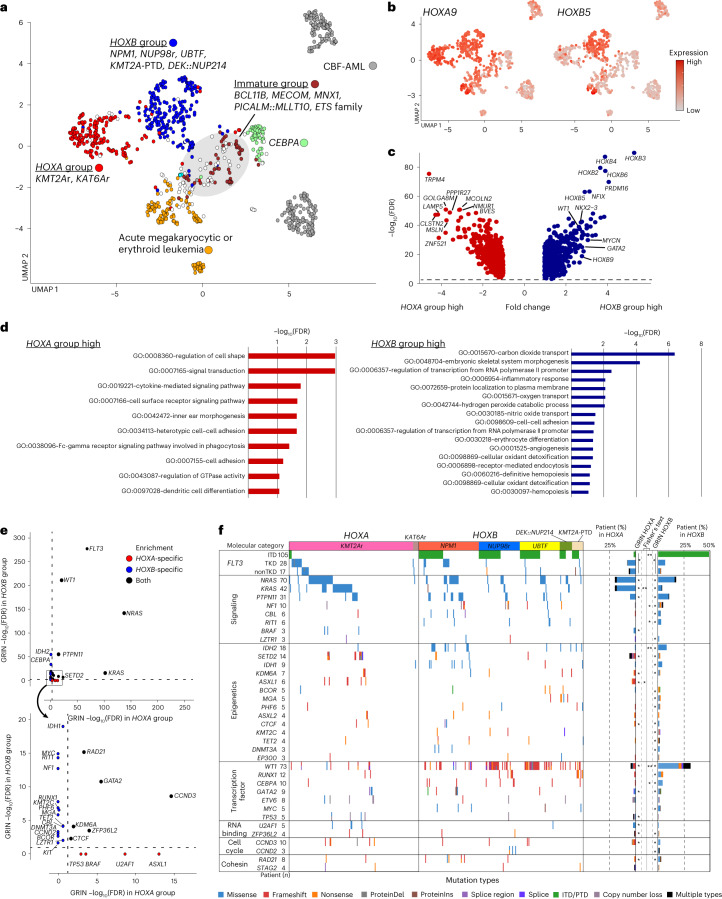
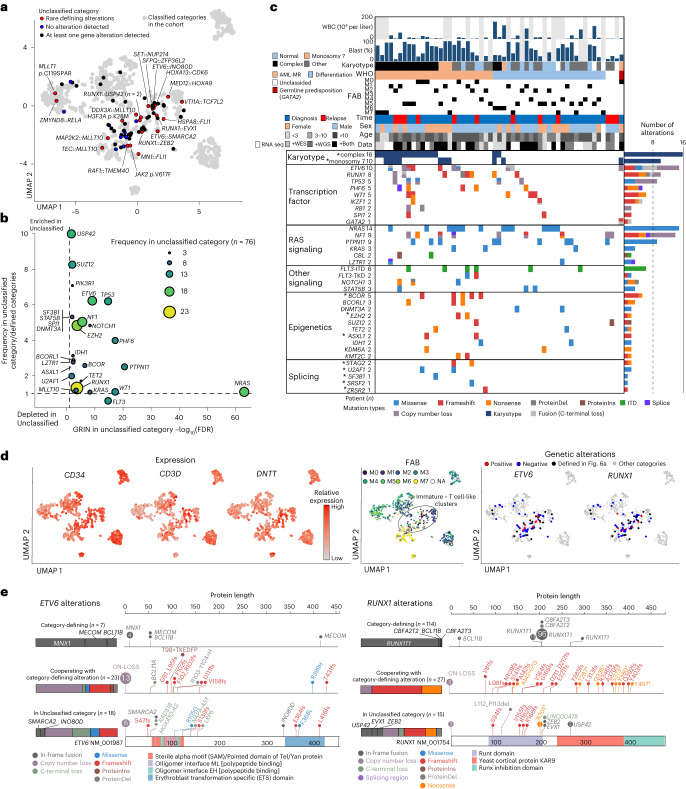
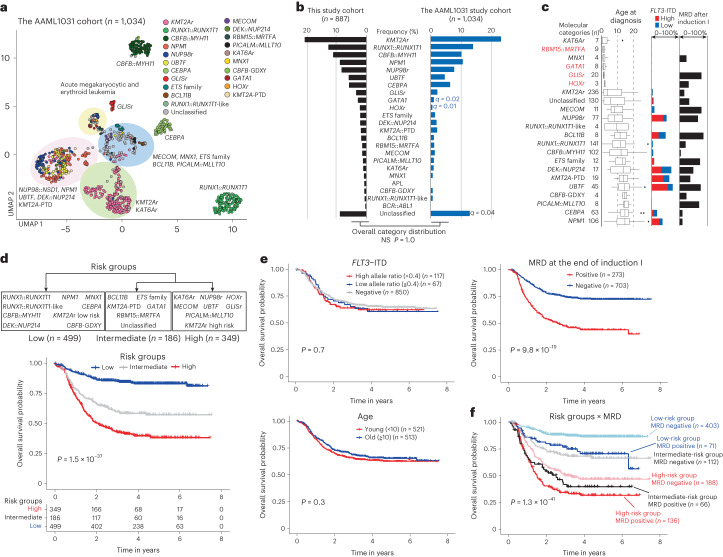
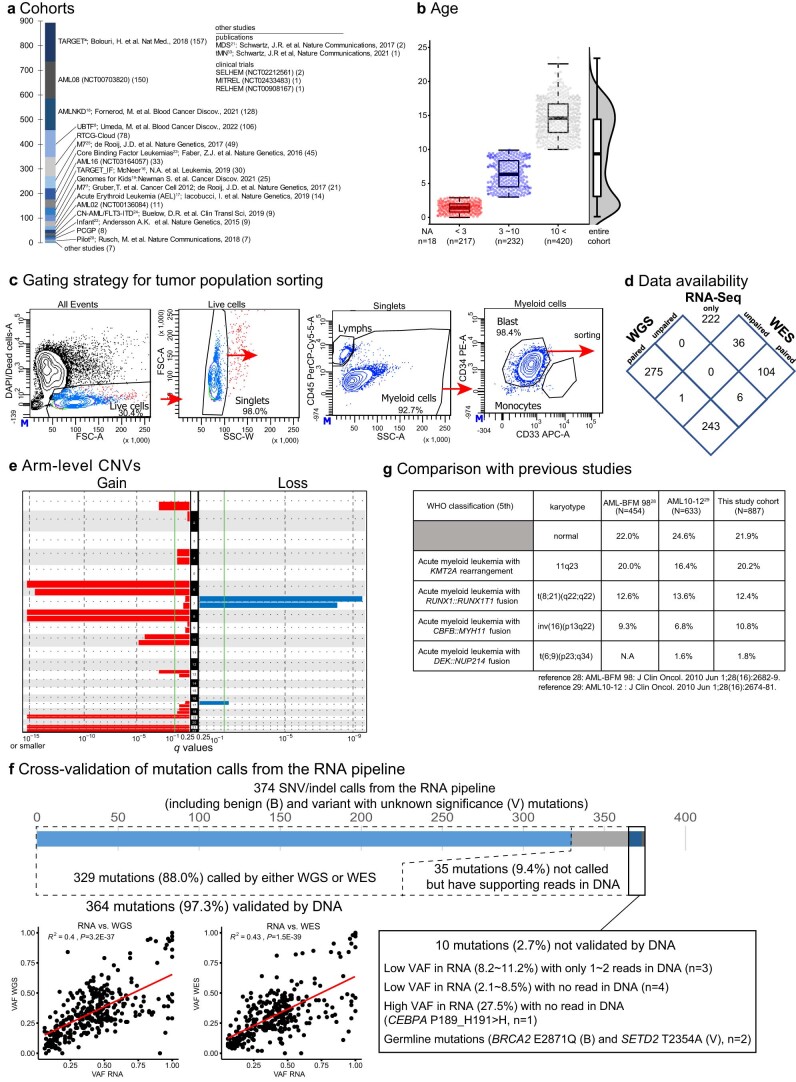
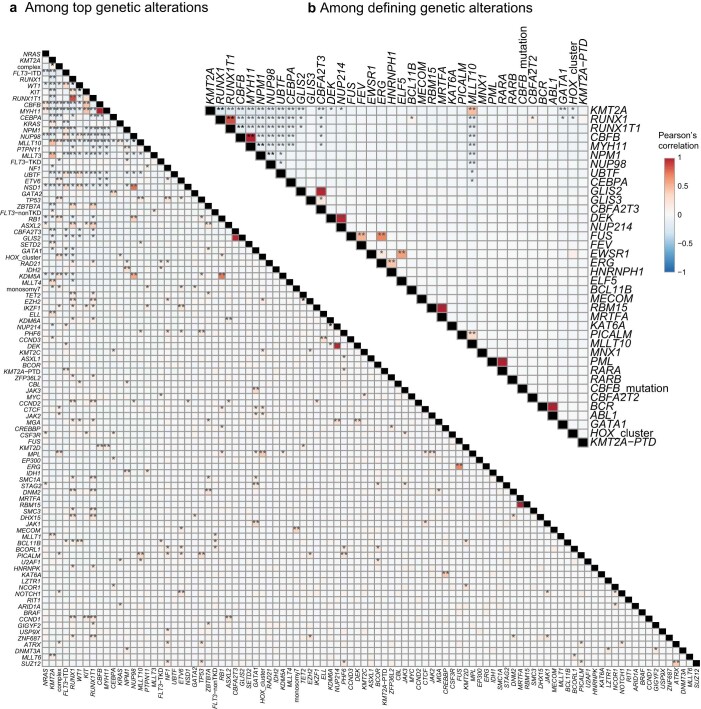
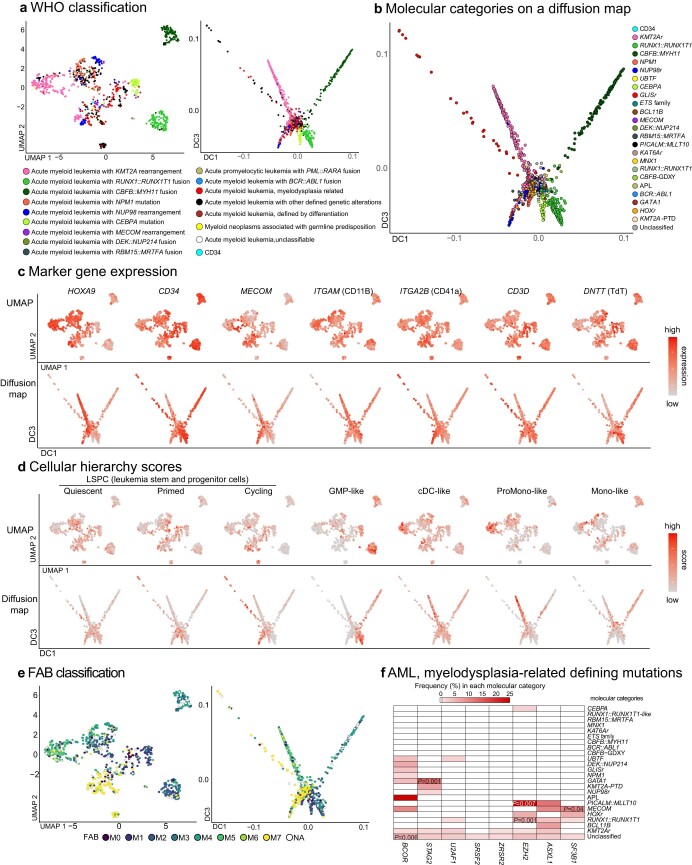
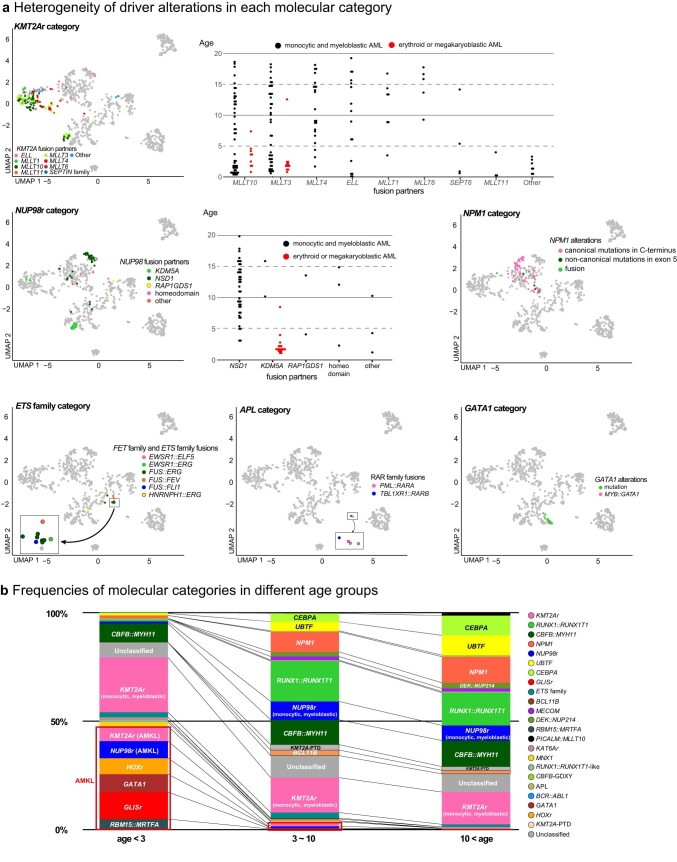
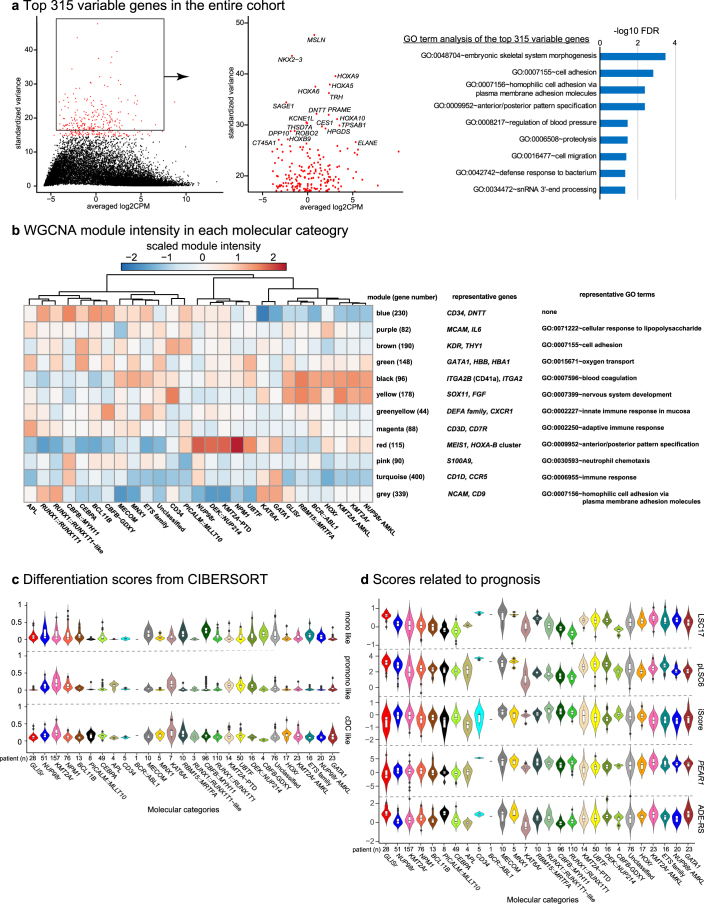
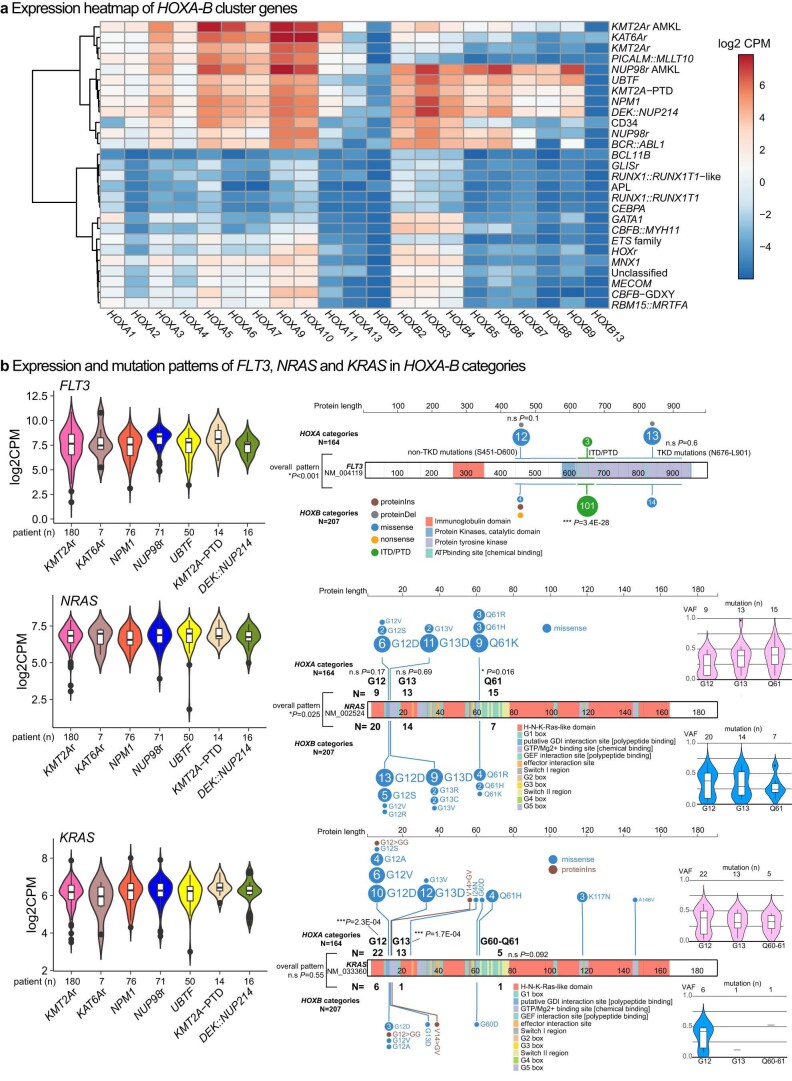
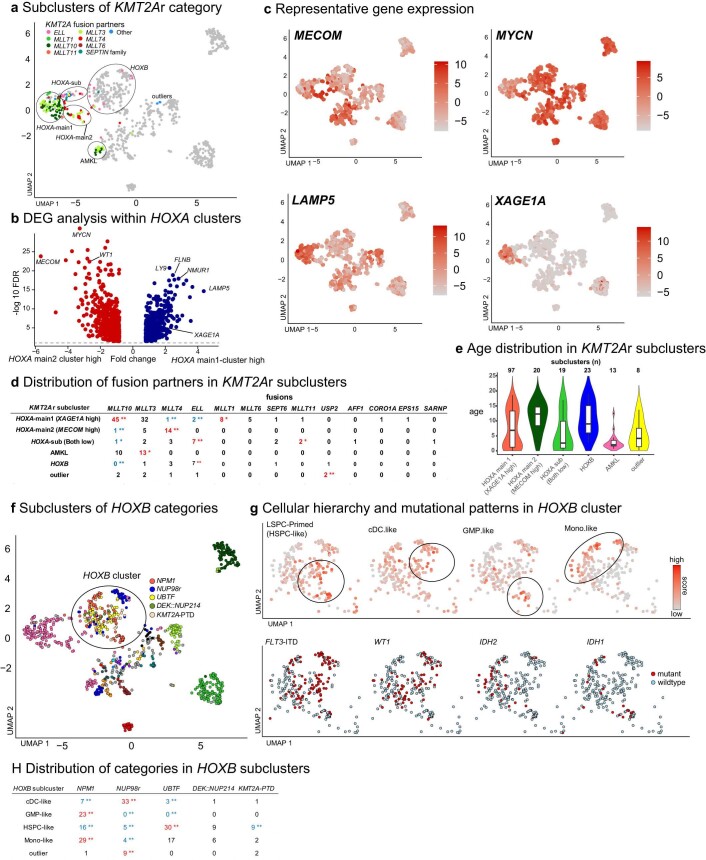
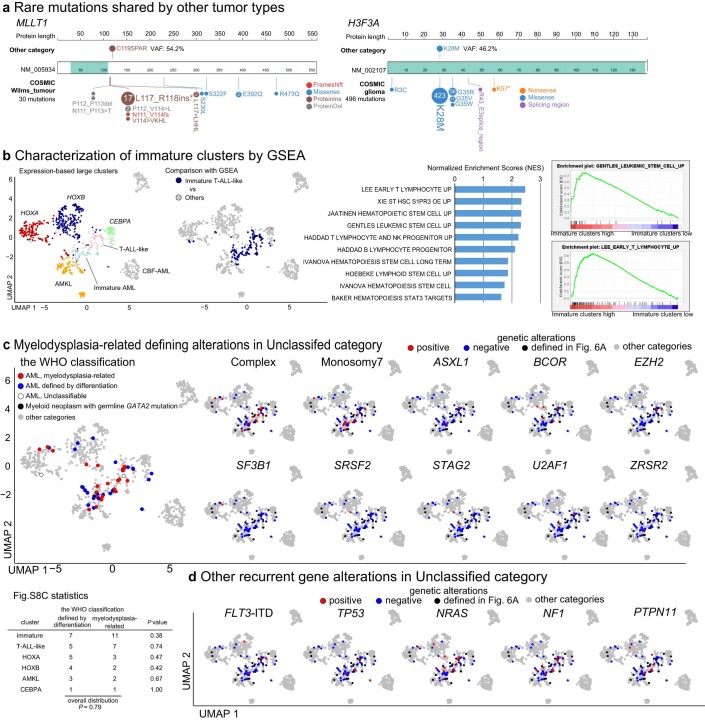
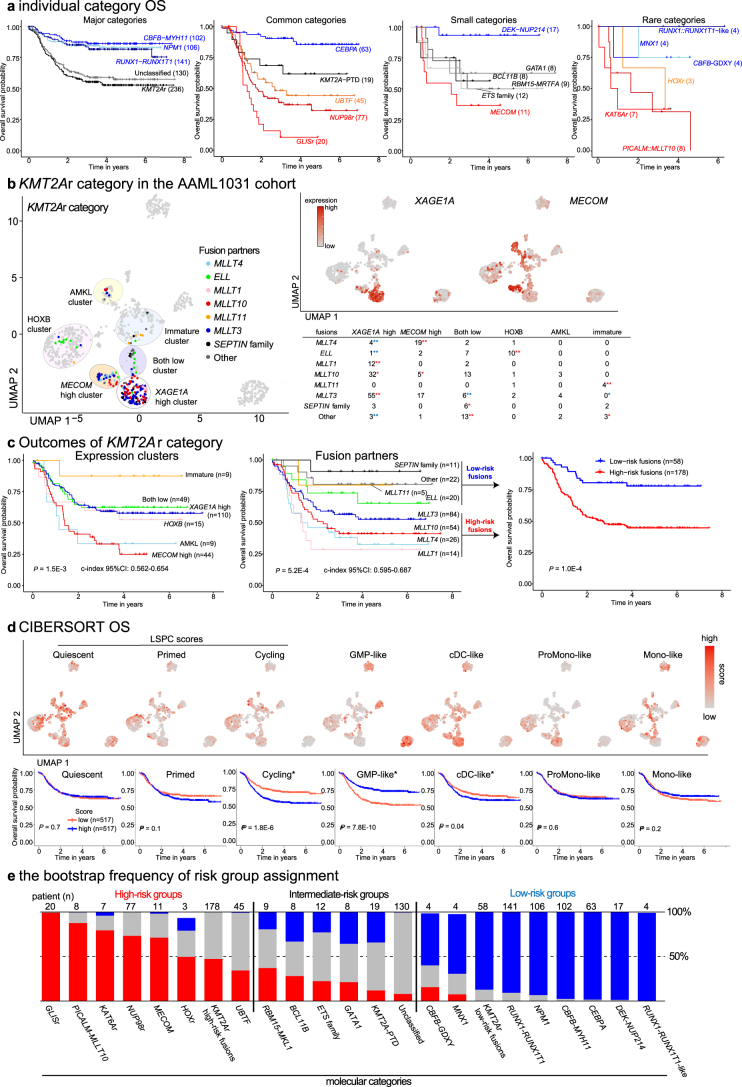
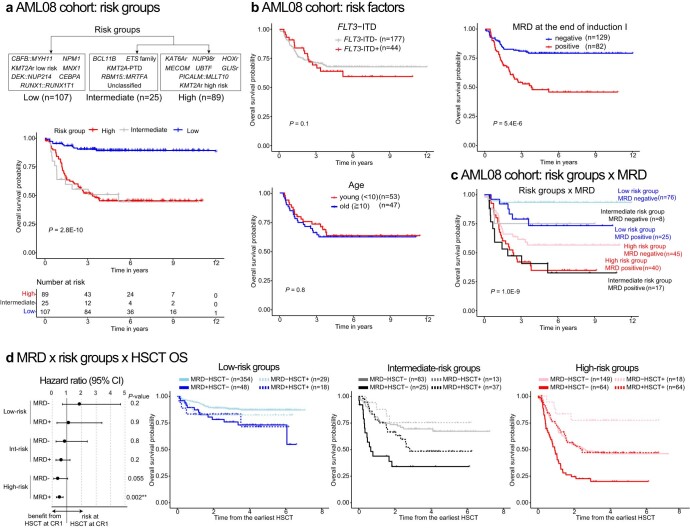
Recent studies on pediatric acute myeloid leukemia (pAML) have revealed pediatric-specific driver alterations, many of which are underrepresented in the current classification schemas. To comprehensively define the genomic landscape of pAML, we systematically categorized 887 pAML into 23 mutually distinct molecular categories, including new major entities such as UBTF or BCL11B, covering 91.4% of the cohort. These molecular categories were associated with unique expression profiles and mutational patterns. For instance, molecular categories characterized by specific HOXA or HOXB expression signatures showed distinct mutation patterns of RAS pathway genes, FLT3 or WT1, suggesting shared biological mechanisms. We show that molecular categories were strongly associated with clinical outcomes using two independent cohorts, leading to the establishment of a new prognostic framework for pAML based on these updated molecular categories and minimal residual disease. Together, this comprehensive diagnostic and prognostic framework forms the basis for future classification of pAML and treatment strategies.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Update of
-
Proposal of a new genomic framework for categorization of pediatric acute myeloid leukemia associated with prognosis.Res Sq [Preprint]. 2023 May 29:rs.3.rs-2925426. doi: 10.21203/rs.3.rs-2925426/v1. Res Sq. 2023. Update in: Nat Genet. 2024 Feb;56(2):281-293. doi: 10.1038/s41588-023-01640-3. PMID: 37398194 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
