

Aberrant DNA repair reveals a vulnerability in histone H3.3-mutant brain tumors
- PMID: 38214234
- PMCID: PMC10954481
- DOI: 10.1093/nar/gkad1257
Aberrant DNA repair reveals a vulnerability in histone H3.3-mutant brain tumors
Abstract
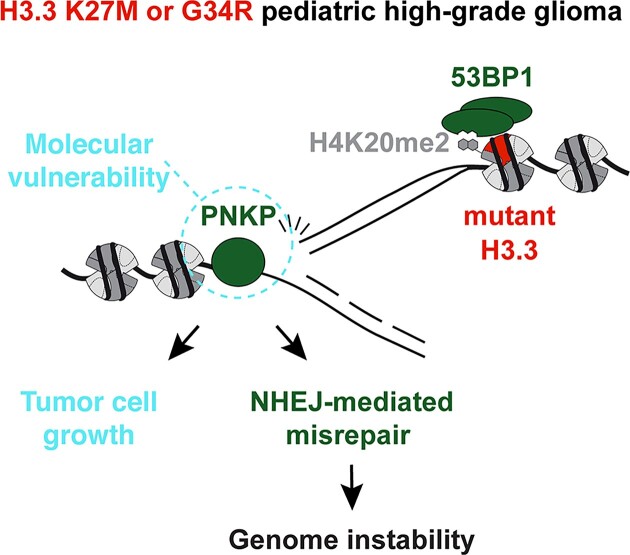
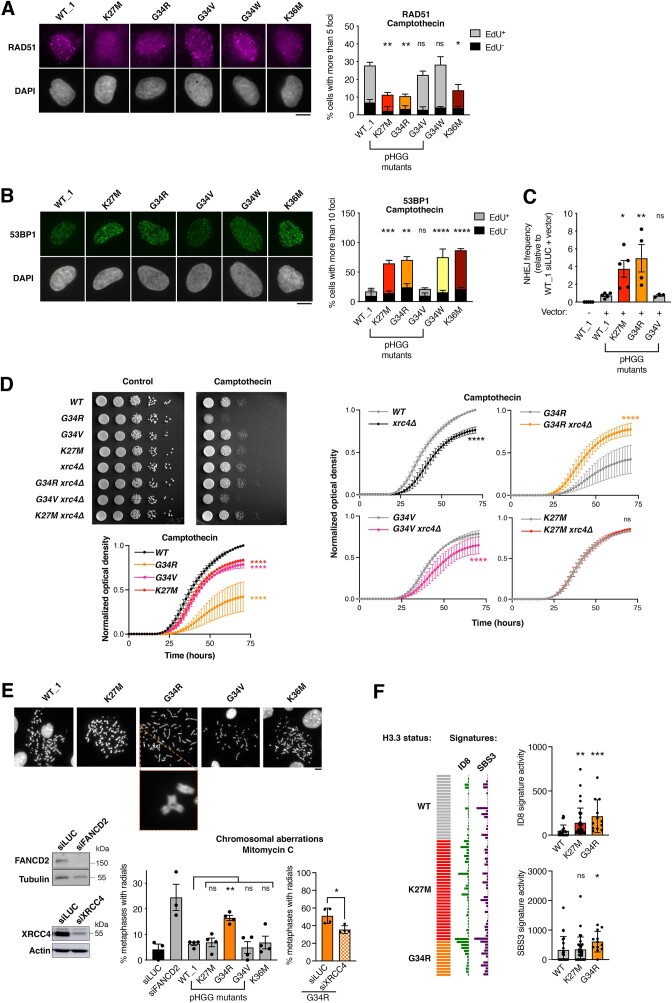
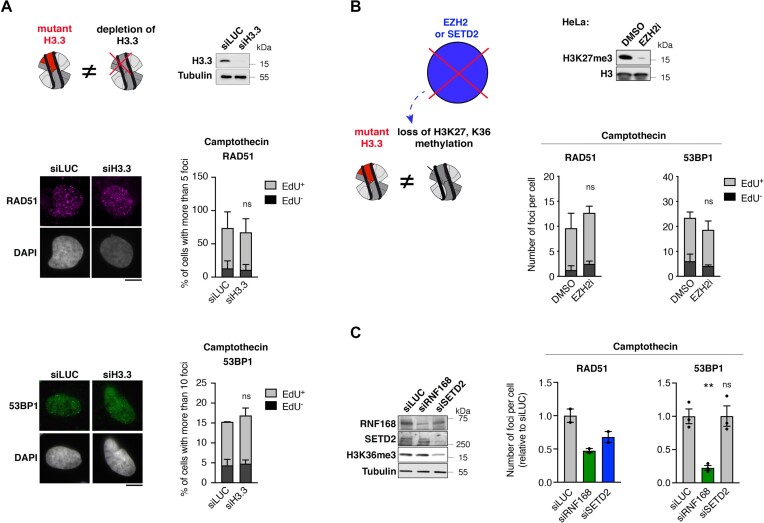
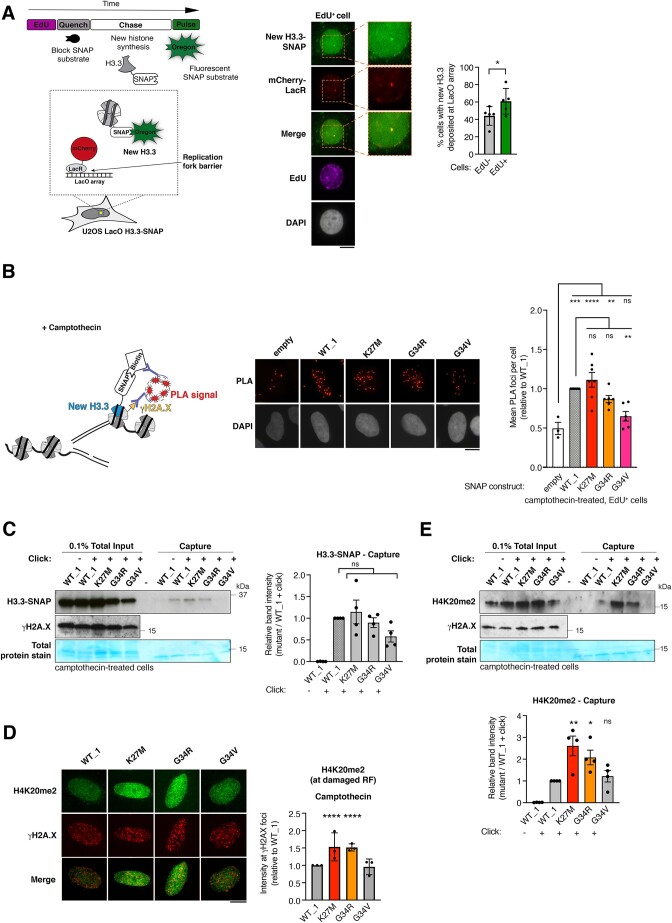
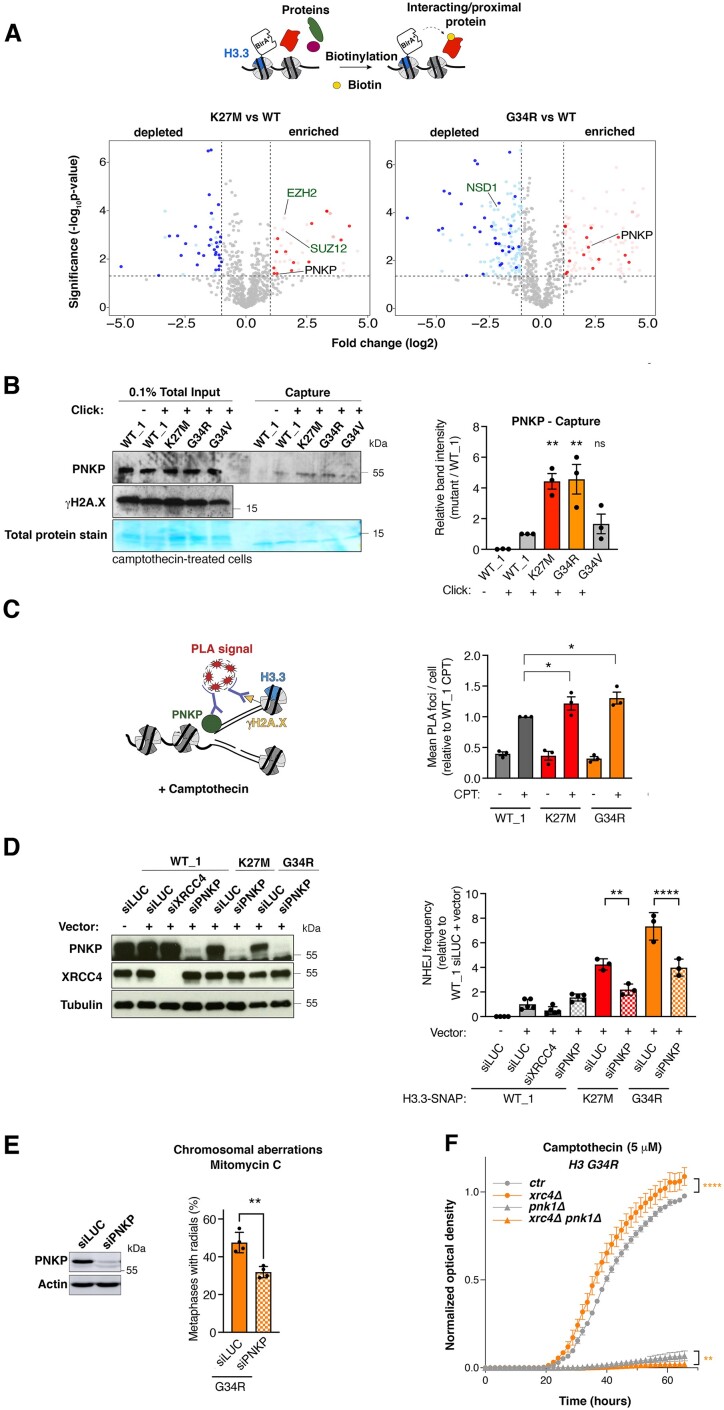
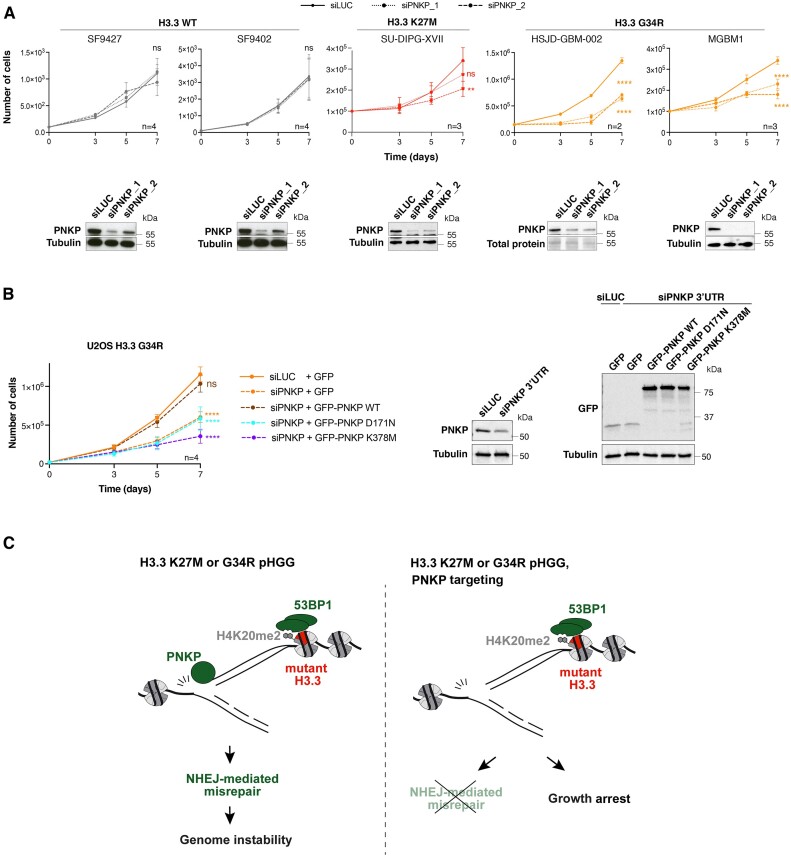
Pediatric high-grade gliomas (pHGG) are devastating and incurable brain tumors with recurrent mutations in histone H3.3. These mutations promote oncogenesis by dysregulating gene expression through alterations of histone modifications. We identify aberrant DNA repair as an independent mechanism, which fosters genome instability in H3.3 mutant pHGG, and opens new therapeutic options. The two most frequent H3.3 mutations in pHGG, K27M and G34R, drive aberrant repair of replication-associated damage by non-homologous end joining (NHEJ). Aberrant NHEJ is mediated by the DNA repair enzyme polynucleotide kinase 3'-phosphatase (PNKP), which shows increased association with mutant H3.3 at damaged replication forks. PNKP sustains the proliferation of cells bearing H3.3 mutations, thus conferring a molecular vulnerability, specific to mutant cells, with potential for therapeutic targeting.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Schwartzentruber J., Korshunov A., Liu X.-Y., Jones D.T.W., Pfaff E., Jacob K., Sturm D., Fontebasso A.M., Quang D.-A.K., Tönjes M. et al. . Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226–231. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
