

Integrated proteogenomic characterization of glioblastoma evolution
- PMID: 38215747
- PMCID: PMC10939876
- DOI: 10.1016/j.ccell.2023.12.015
Integrated proteogenomic characterization of glioblastoma evolution
Abstract
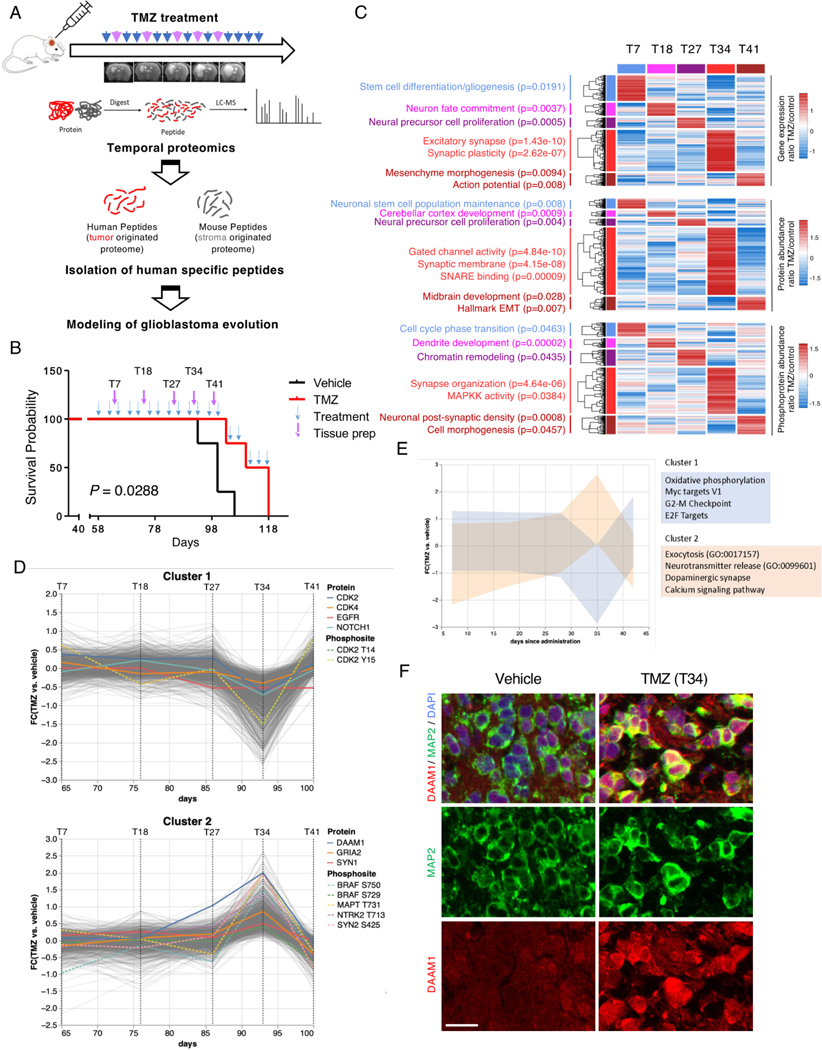
The evolutionary trajectory of glioblastoma (GBM) is a multifaceted biological process that extends beyond genetic alterations alone. Here, we perform an integrative proteogenomic analysis of 123 longitudinal glioblastoma pairs and identify a highly proliferative cellular state at diagnosis and replacement by activation of neuronal transition and synaptogenic pathways in recurrent tumors. Proteomic and phosphoproteomic analyses reveal that the molecular transition to neuronal state at recurrence is marked by post-translational activation of the wingless-related integration site (WNT)/ planar cell polarity (PCP) signaling pathway and BRAF protein kinase. Consistently, multi-omic analysis of patient-derived xenograft (PDX) models mirror similar patterns of evolutionary trajectory. Inhibition of B-raf proto-oncogene (BRAF) kinase impairs both neuronal transition and migration capability of recurrent tumor cells, phenotypic hallmarks of post-therapy progression. Combinatorial treatment of temozolomide (TMZ) with BRAF inhibitor, vemurafenib, significantly extends the survival of PDX models. This study provides comprehensive insights into the biological mechanisms of glioblastoma evolution and treatment resistance, highlighting promising therapeutic strategies for clinical intervention.
Keywords: BRAF; longitudinal glioblastoma; neuronal; proteogenomics; recurrence; synapse.
Copyright © 2023 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures







References
-
- Fougner V, Hasselbalch B, Lassen U, Weischenfeldt J, Poulsen HS, and Urup T. (2022). Implementing targeted therapies in the treatment of glioblastoma: Previous shortcomings, future promises, and a multimodal strategy recommendation. Neurooncol Adv 4, vdac157. 10.1093/noajnl/vdac157. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
