

Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching
- PMID: 38225394
- PMCID: PMC11053127
- DOI: 10.1038/s41401-023-01222-3
Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching
Abstract
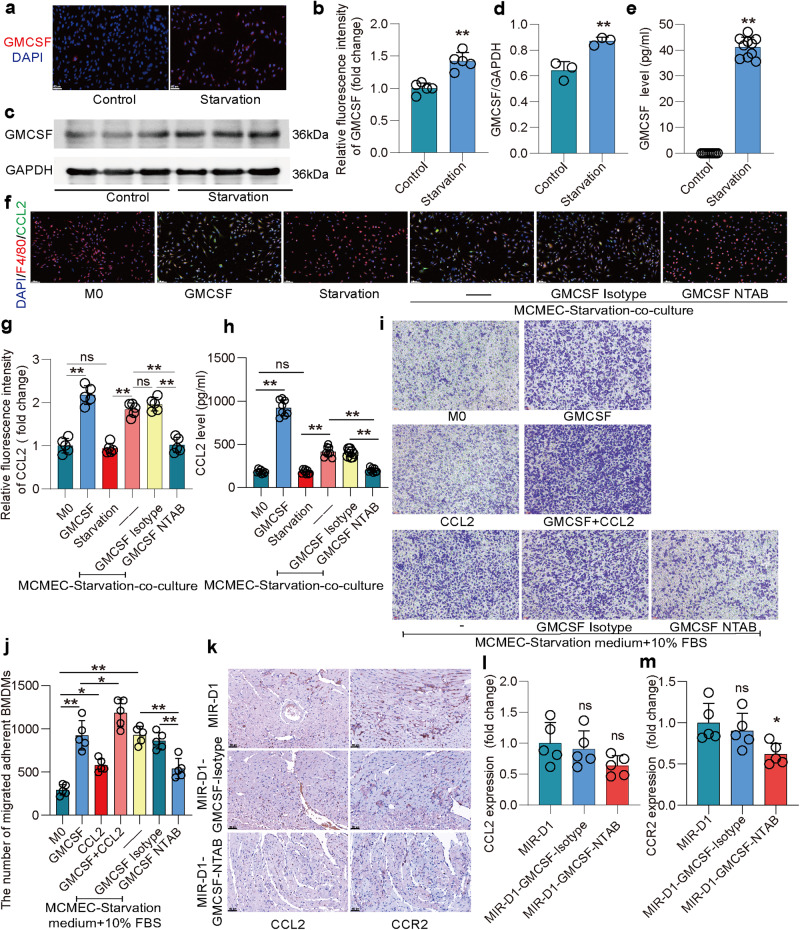
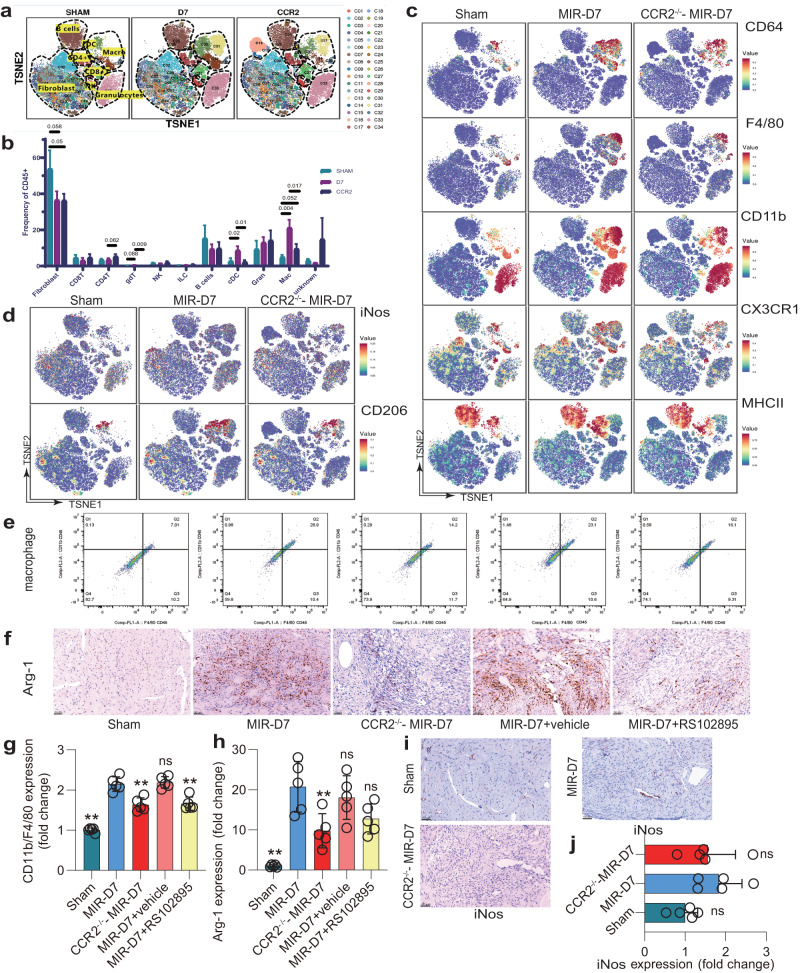
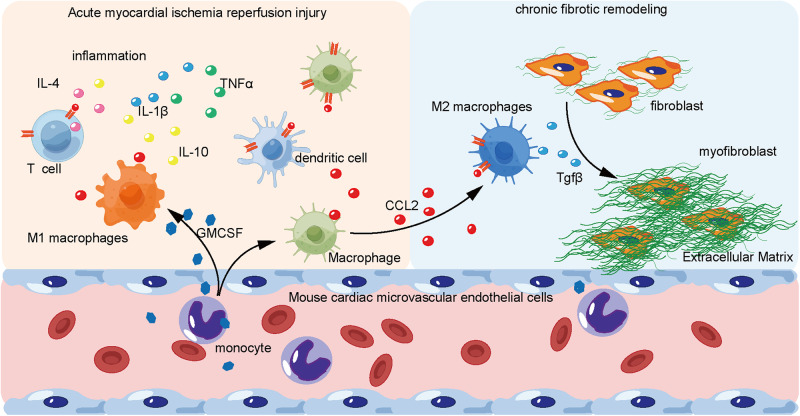
Following acute myocardial ischemia reperfusion (MIR), macrophages infiltrate damaged cardiac tissue and alter their polarization phenotype to respond to acute inflammation and chronic fibrotic remodeling. In this study we investigated the role of macrophages in post-ischemic myocardial fibrosis and explored therapeutic targets for myocardial fibrosis. Male mice were subjected to ligation of the left coronary artery for 30 min. We first detected the levels of chemokines in heart tissue that recruited immune cells infiltrating into the heart, and found that granulocyte-macrophage colony-stimulating factor (GMCSF) released by mouse cardiac microvascular endothelial cells (MCMECs) peaked at 6 h after reperfusion, and c-c motif chemokine ligand 2 (CCL2) released by GMCSF-induced macrophages peaked at 24 h after reperfusion. In co-culture of BMDMs with MCMECs, we demonstrated that GMCSF derived from MCMECs stimulated the release of CCL2 by BMDMs and effectively promoted the migration of BMDMs. We also confirmed that GMCSF promoted M1 polarization of macrophages in vitro, while GMCSF neutralizing antibodies (NTABs) blocked CCL2/CCR2 signaling. In MIR mouse heart, we showed that GMCSF activated CCL2/CCR2 signaling to promote NLRP3/caspase-1/IL-1β-mediated and amplified inflammatory damage. Knockdown of CC chemokine receptor 2 gene (CCR2-/-), or administration of specific CCR2 inhibitor RS102895 (5 mg/kg per 12 h, i.p., one day before MIR and continuously until the end of the experiment) effectively reduced the area of myocardial infarction, and down-regulated inflammatory mediators and NLRP3/Caspase-1/IL-1β signaling. Mass cytometry confirmed that M2 macrophages played an important role during fibrosis, while macrophage-depleted mice exhibited significantly reduced transforming growth factor-β (Tgf-β) levels in heart tissue after MIR. In co-culture of macrophages with fibroblasts, treatment with recombinant mouse CCL2 stimulated macrophages to release a large amount of Tgf-β, and promoted the release of Col1α1 by fibroblasts. This effect was diminished in BMDMs from CCR2-/- mice. After knocking out or inhibiting CCR2-gene, the levels of Tgf-β were significantly reduced, as was the level of myocardial fibrosis, and cardiac function was protected. This study confirms that the acute injury to chronic fibrosis transition after MIR in mice is mediated by GMCSF/CCL2/CCR2 signaling in macrophages through NLRP3 inflammatory cascade and the phenotype switching.
Keywords: CCL2/CCR2; GMCSF; cardiac fibrosis; macrophages polarization; myocardial ischemia reperfusion; transforming growth factor-β.
© 2024. The Author(s), under exclusive licence to Shanghai Institute of Materia Medica, Chinese Academy of Sciences and Chinese Pharmacological Society.
Conflict of interest statement
The authors declare no competing interests.
Figures






References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
