

Glucocorticoid receptor-dependent therapeutic efficacy of tauroursodeoxycholic acid in preclinical models of spinocerebellar ataxia type 3
- PMID: 38227368
- PMCID: PMC10904051
- DOI: 10.1172/JCI162246
Glucocorticoid receptor-dependent therapeutic efficacy of tauroursodeoxycholic acid in preclinical models of spinocerebellar ataxia type 3
Abstract
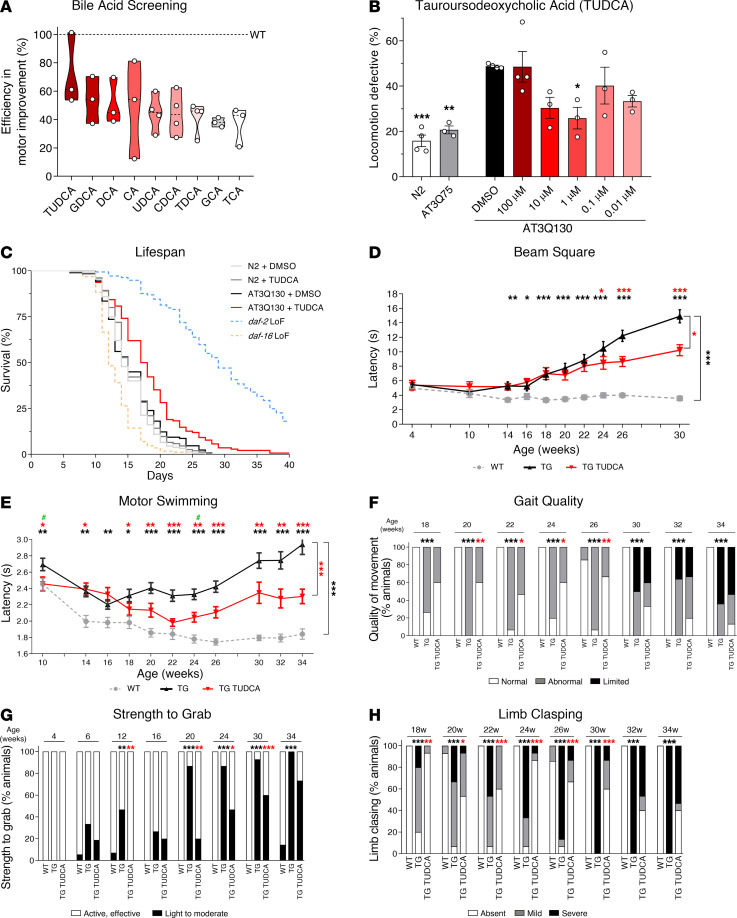
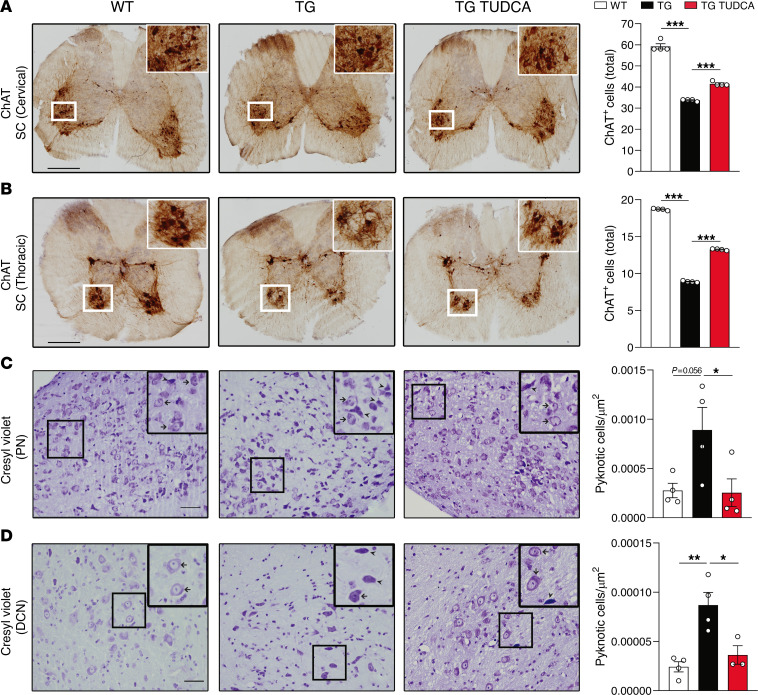
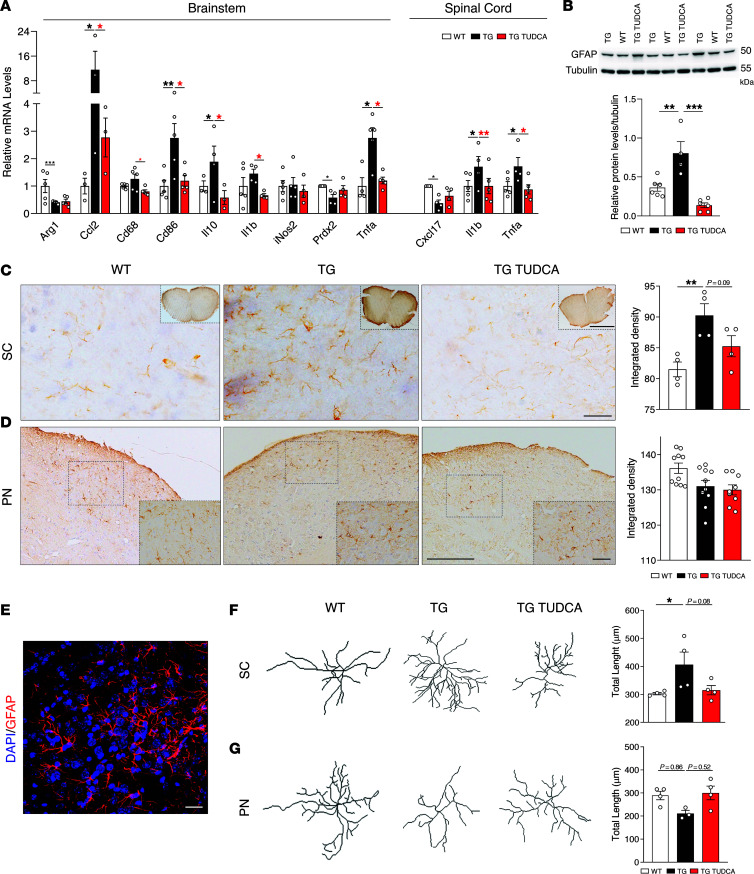
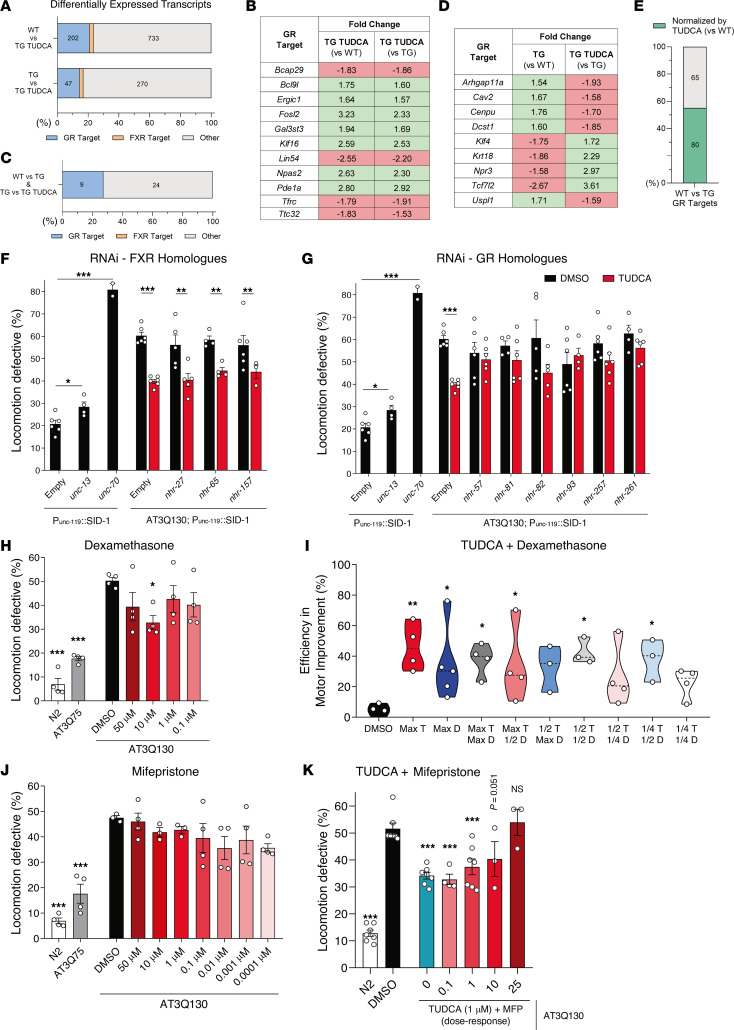
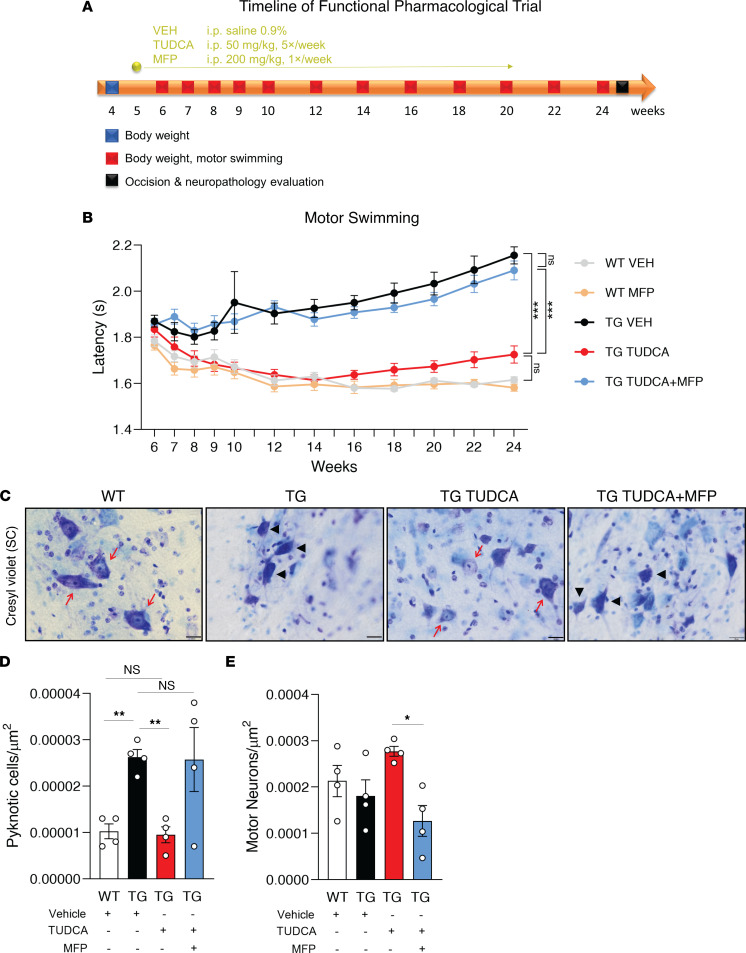
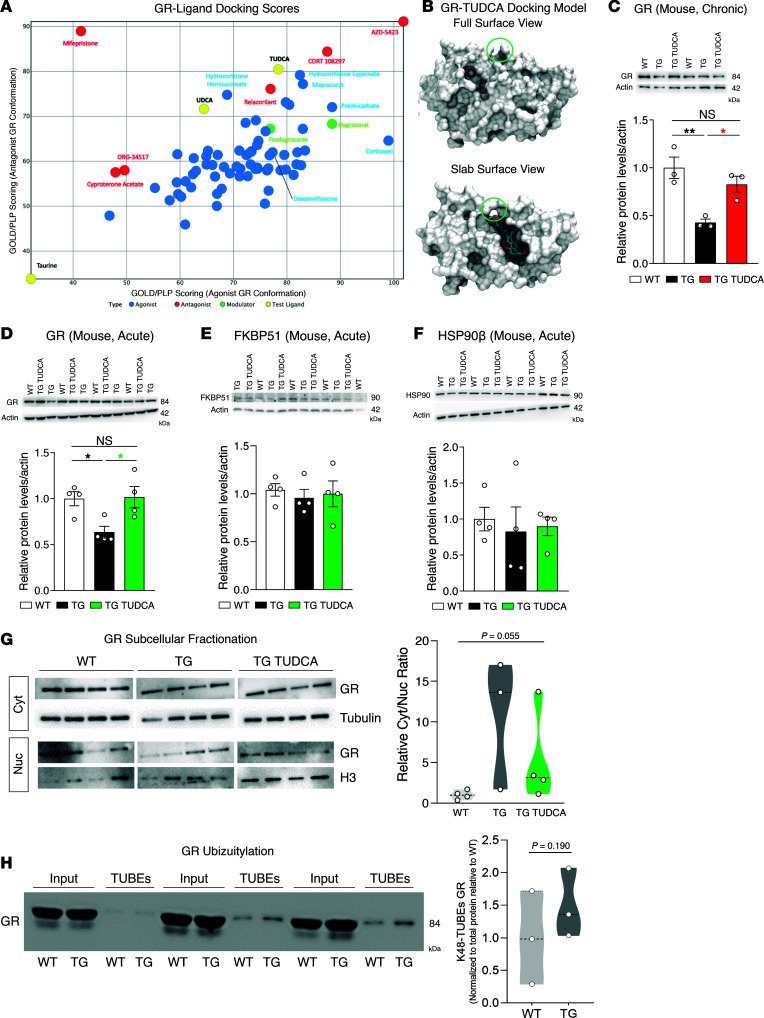
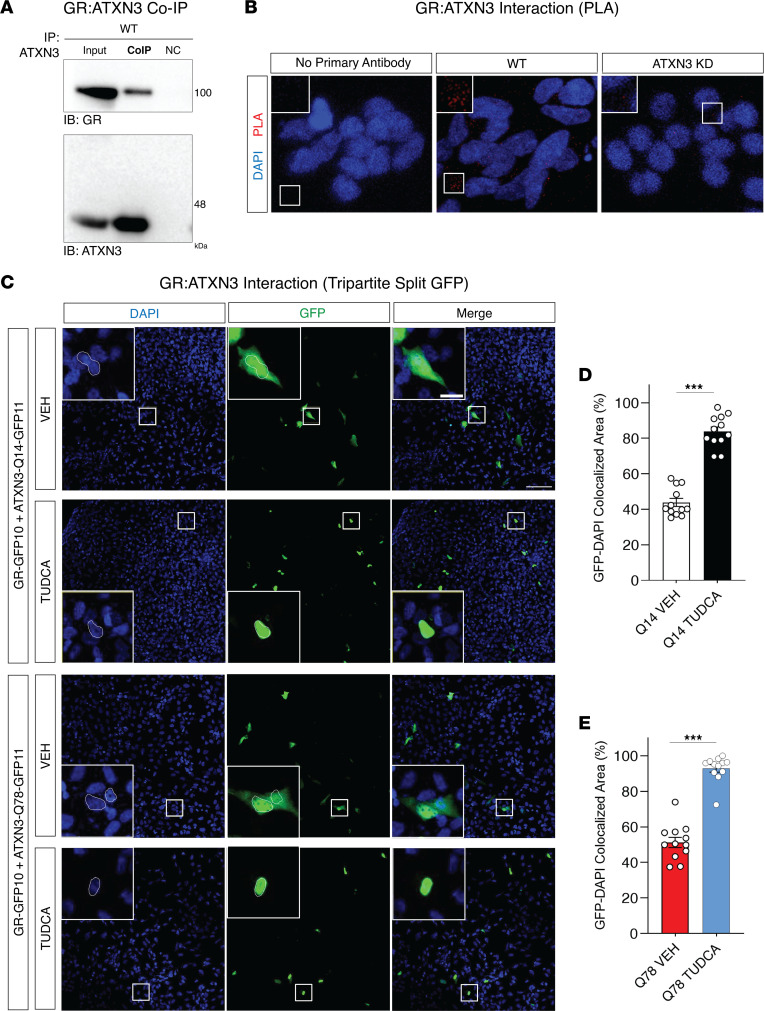
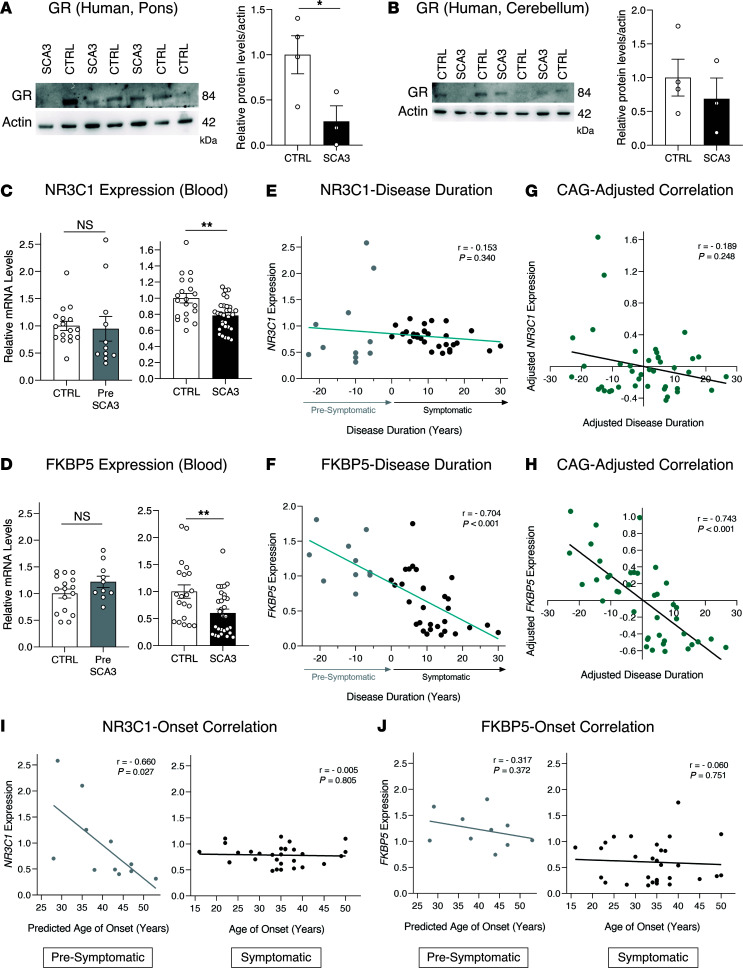
Spinocerebellar ataxia type 3 (SCA3) is an adult-onset neurodegenerative disease caused by a polyglutamine expansion in the ataxin-3 (ATXN3) gene. No effective treatment is available for this disorder, other than symptom-directed approaches. Bile acids have shown therapeutic efficacy in neurodegenerative disease models. Here, we pinpointed tauroursodeoxycholic acid (TUDCA) as an efficient therapeutic, improving the motor and neuropathological phenotype of SCA3 nematode and mouse models. Surprisingly, transcriptomic and functional in vivo data showed that TUDCA acts in neuronal tissue through the glucocorticoid receptor (GR), but independently of its canonical receptor, the farnesoid X receptor (FXR). TUDCA was predicted to bind to the GR, in a similar fashion to corticosteroid molecules. GR levels were decreased in disease-affected brain regions, likely due to increased protein degradation as a consequence of ATXN3 dysfunction being restored by TUDCA treatment. Analysis of a SCA3 clinical cohort showed intriguing correlations between the peripheral expression of GR and the predicted age at disease onset in presymptomatic subjects and FKBP5 expression with disease progression, suggesting this pathway as a potential source of biomarkers for future study. We have established a novel in vivo mechanism for the neuroprotective effects of TUDCA in SCA3 and propose this readily available drug for clinical trials in SCA3 patients.
Keywords: Drug therapy; Genetic diseases; Molecular biology; Neuroscience; Therapeutics.
Figures
Similar articles
-
Impaired Oligodendrocyte Maturation Is an Early Feature in SCA3 Disease Pathogenesis.J Neurosci. 2022 Feb 23;42(8):1604-1617. doi: 10.1523/JNEUROSCI.1954-20.2021. Epub 2022 Jan 18. J Neurosci. 2022. PMID: 35042771 Free PMC article.
-
In vivo assessment of riluzole as a potential therapeutic drug for spinocerebellar ataxia type 3.J Neurochem. 2016 Jul;138(1):150-62. doi: 10.1111/jnc.13606. J Neurochem. 2016. PMID: 26990650
-
Treatment with sodium butyrate induces autophagy resulting in therapeutic benefits for spinocerebellar ataxia type 3.FASEB J. 2024 Jan 31;38(2):e23429. doi: 10.1096/fj.202300963RR. FASEB J. 2024. PMID: 38258931
-
Autophagy in Spinocerebellar Ataxia Type 3: From Pathogenesis to Therapeutics.Int J Mol Sci. 2023 Apr 17;24(8):7405. doi: 10.3390/ijms24087405. Int J Mol Sci. 2023. PMID: 37108570 Free PMC article. Review.
-
From Pathogenesis to Novel Therapeutics for Spinocerebellar Ataxia Type 3: Evading Potholes on the Way to Translation.Neurotherapeutics. 2019 Oct;16(4):1009-1031. doi: 10.1007/s13311-019-00798-1. Neurotherapeutics. 2019. PMID: 31691128 Free PMC article. Review.
Cited by
-
Prediction of protein interactions with function in protein (de-)phosphorylation.PLoS One. 2025 Mar 3;20(3):e0319084. doi: 10.1371/journal.pone.0319084. eCollection 2025. PLoS One. 2025. PMID: 40029919 Free PMC article.
-
From stigma to increased social acceptance? Living with Machado-Joseph disease in São Miguel, Azores, Portugal.J Community Genet. 2024 Aug 31. doi: 10.1007/s12687-024-00731-w. Online ahead of print. J Community Genet. 2024. PMID: 39215935
-
Tauroursodeoxycholic acid targets HSP90 to promote protein homeostasis and extends healthy lifespan.Sci China Life Sci. 2025 Feb;68(2):416-430. doi: 10.1007/s11427-024-2717-6. Epub 2024 Sep 24. Sci China Life Sci. 2025. PMID: 39327392
-
Bile acids target an exposed cavity in the glucocorticoid receptor modulating receptor self-assembly, chromatin binding and transcriptional activity.bioRxiv [Preprint]. 2025 May 16:2025.05.13.653693. doi: 10.1101/2025.05.13.653693. bioRxiv. 2025. PMID: 40463087 Free PMC article. Preprint.
-
Emerging Roles of Bile Acids and TGR5 in the Central Nervous System: Molecular Functions and Therapeutic Implications.Int J Mol Sci. 2024 Aug 27;25(17):9279. doi: 10.3390/ijms25179279. Int J Mol Sci. 2024. PMID: 39273226 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
