

Clinicopathological correlates in the frontotemporal lobar degeneration-motor neuron disease spectrum
- PMID: 38227807
- PMCID: PMC11224598
- DOI: 10.1093/brain/awae011
Clinicopathological correlates in the frontotemporal lobar degeneration-motor neuron disease spectrum
Abstract
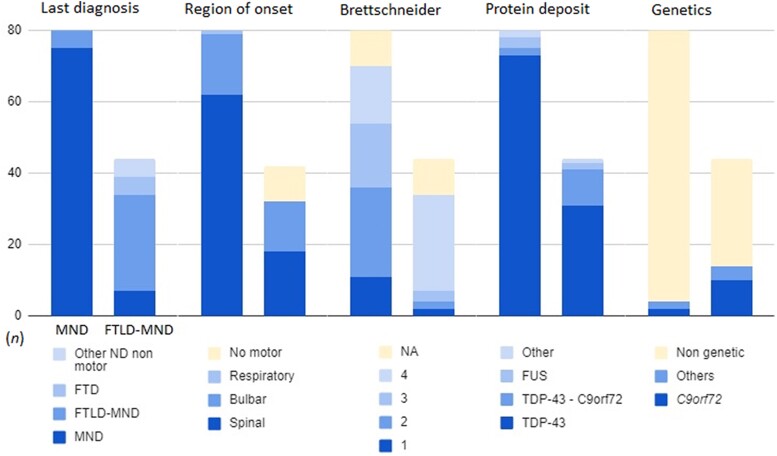
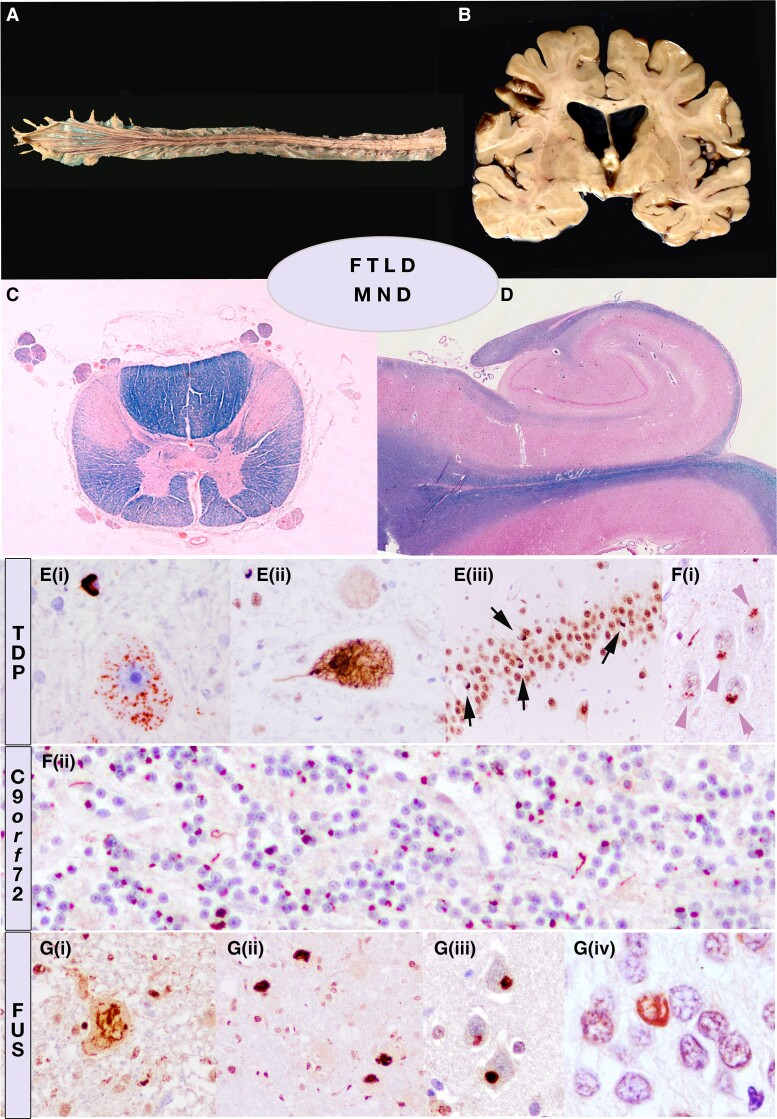
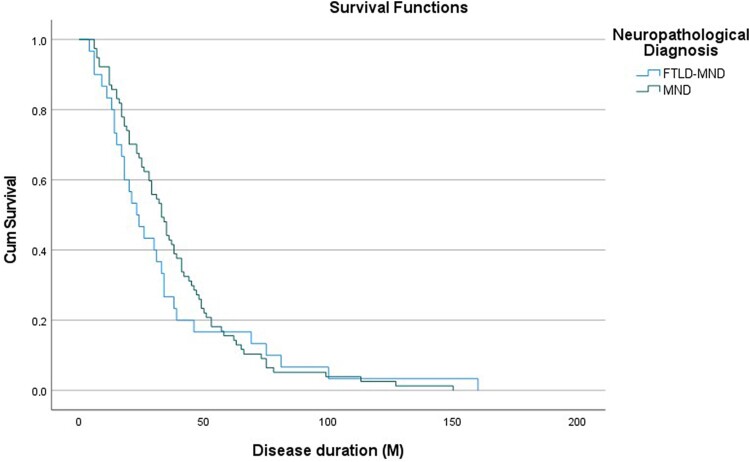
Amyotrophic lateral sclerosis (ALS) is a devastating motor neuron disease (MND) that shares a common clinical, genetic and pathologic spectrum with frontotemporal dementia (FTD). It is highly heterogeneous in its presentation and features. Up to 50% of patients with MND develop cognitive-behavioural symptoms during the course of the disease, meeting criteria for FTD in 10%-15% of cases. In the absence of a precise biomarker, neuropathology is still a valuable tool to understand disease nosology, reach a definite diagnostic confirmation and help define specific subgroups of patients with common phenotypic, genetic and biomarker profiles. However, few neuropathological series have been published, and the frequency of frontotemporal lobar degeneration (FTLD) in MND is difficult to estimate. In this work we describe a large clinicopathological series of MND patients, analysing the frequency of concurrent FTLD changes and trying to define specific subgroups of patients based on their clinical, genetic and pathological characteristics. We performed an observational, retrospective, multicentre case study. We included all cases meeting neuropathological criteria for MND from the Neurological Tissue Bank of the FRCB-IDIBAPS-Hospital Clínic Barcelona Biobank between 1994 and 2022, regardless of their last clinical diagnosis. While brain donation is encouraged in all patients, it is performed in very few, and representativeness of the cohort might not be precise for all patients with MND. We retrospectively reviewed clinical and neuropathological data and describe the main clinical, genetic and pathogenic features, comparing neuropathologic groups between MND with and without FTLD changes and aiming to define specific subgroups. We included brain samples from 124 patients, 44 of whom (35.5%) had FTLD neuropathologic features (i.e. FTLD-MND). Pathologic TDP-43 aggregates were present in 93.6% of the cohort and were more extensive (higher Brettschneider stage) in those with concurrent FTLD (P < 0.001). Motor symptom onset was more frequent in the bulbar region in FTLD-MND cases than in those with isolated MND (P = 0.023), with no differences in survival. We observed a better clinicopathological correlation in the MND group than in the FTLD-MND group (93.8% versus 61.4%; P < 0.001). Pathogenic genetic variants were more common in the FTLD-MND group, especially C9orf72. We describe a frequency of FTLD of 35.5% in our series of neuropathologically confirmed cases of MND. The FTLD-MND spectrum is highly heterogeneous in all aspects, especially in patients with FTLD, in whom it is particularly difficult to define specific subgroups. In the absence of definite biomarkers, neuropathology remains a valuable tool for a definite diagnosis, increasing our knowledge in disease nosology.
Keywords: amyotrophic lateral sclerosis; frontotemporal dementia; motor neuron disease; neuropathology.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures
References
-
- van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:2084–2098. - PubMed
-
- Phukan J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: A population-based study. J Neurol Neurosurg Psychiatry. 2012;83:102–108. - PubMed
-
- Elamin M, Phukan J, Bede P, et al. Executive dysfunction is a negative prognostic indicator in patients with ALS without dementia. Neurology. 2011;76:1263–1269. - PubMed
-
- Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134:2582–2594. - PubMed
-
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
