

The impact of damaging epilepsy and cardiac genetic variant burden in sudden death in the young
- PMID: 38229148
- PMCID: PMC10792876
- DOI: 10.1186/s13073-024-01284-w
The impact of damaging epilepsy and cardiac genetic variant burden in sudden death in the young
Abstract
Background: Sudden unexpected death in children is a tragic event. Understanding the genetics of sudden death in the young (SDY) enables family counseling and cascade screening. The objective of this study was to characterize genetic variation in an SDY cohort using whole genome sequencing.
Methods: The SDY Case Registry is a National Institutes of Health/Centers for Disease Control and Prevention surveillance effort to discern the prevalence, causes, and risk factors for SDY. The SDY Case Registry prospectively collected clinical data and DNA biospecimens from SDY cases < 20 years of age. SDY cases were collected from medical examiner and coroner offices spanning 13 US jurisdictions from 2015 to 2019. The cohort included 211 children (median age 0.33 year; range 0-20 years), determined to have died suddenly and unexpectedly and from whom DNA biospecimens for DNA extractions and next-of-kin consent were ascertained. A control cohort consisted of 211 randomly sampled, sex- and ancestry-matched individuals from the 1000 Genomes Project. Genetic variation was evaluated in epilepsy, cardiomyopathy, and arrhythmia genes in the SDY and control cohorts. American College of Medical Genetics/Genomics guidelines were used to classify variants as pathogenic or likely pathogenic. Additionally, pathogenic and likely pathogenic genetic variation was identified using a Bayesian-based artificial intelligence (AI) tool.
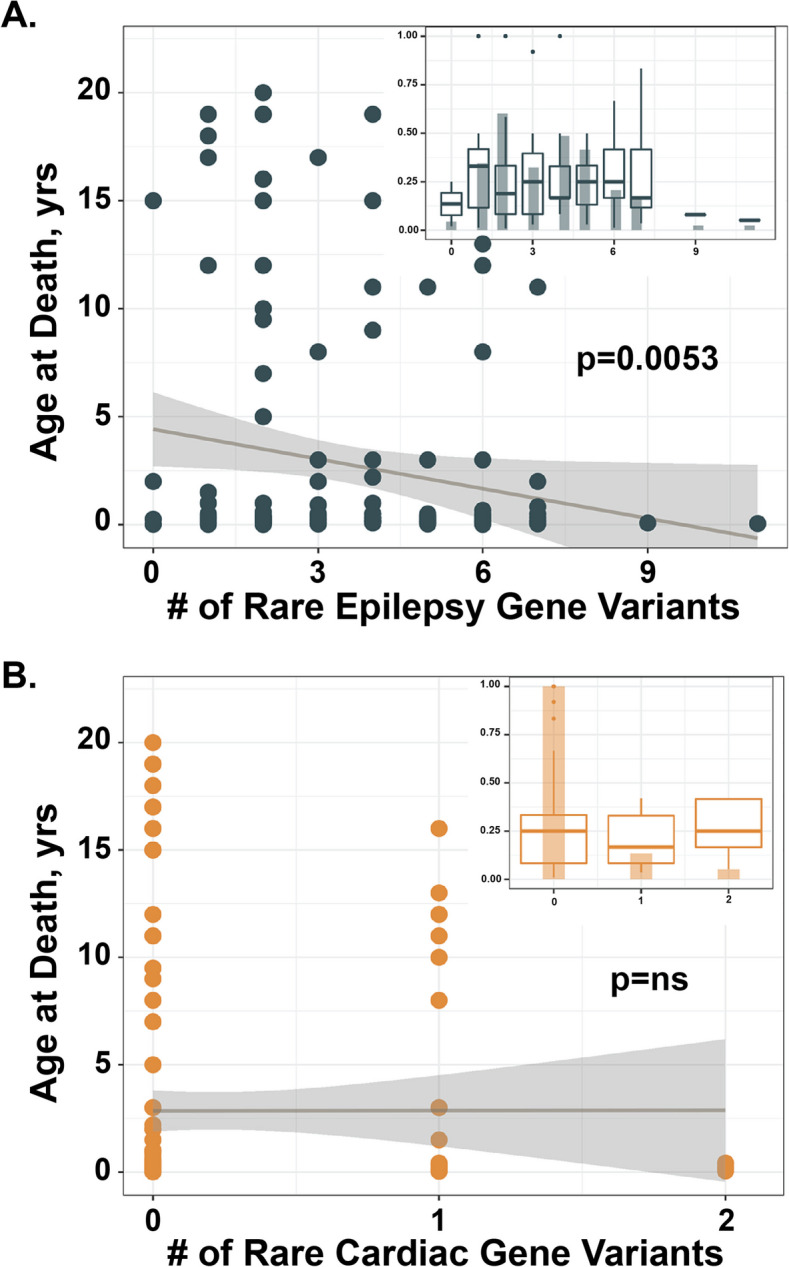
Results: The SDY cohort was 43% European, 29% African, 3% Asian, 16% Hispanic, and 9% with mixed ancestries and 39% female. Six percent of the cohort was found to harbor a pathogenic or likely pathogenic genetic variant in an epilepsy, cardiomyopathy, or arrhythmia gene. The genomes of SDY cases, but not controls, were enriched for rare, potentially damaging variants in epilepsy, cardiomyopathy, and arrhythmia-related genes. A greater number of rare epilepsy genetic variants correlated with younger age at death.
Conclusions: While damaging cardiomyopathy and arrhythmia genes are recognized contributors to SDY, we also observed an enrichment in epilepsy-related genes in the SDY cohort and a correlation between rare epilepsy variation and younger age at death. These findings emphasize the importance of considering epilepsy genes when evaluating SDY.
Keywords: Arrhythmia; Cardiomyopathy; Epilepsy; Gene burden; Genome sequencing; Sudden death in the young.
© 2024. The Author(s).
Conflict of interest statement
MY serves as consultant to Fabric Genomics Inc. and has received consulting fees and stock grants from Fabric Genomics Inc. EMM is a consultant for Amgen, Avidity, AstraZeneca, Cytokinetics, PepGen, Pfizer, Tenaya Therapeutics, Stealth BioTherapeutics, and Invitae and is the founder of Ikaika Therapeutics. The remaining authors declare that they do not have any competing interests.
Figures



Update of
-
The impact of damaging epilepsy and cardiac genetic variant burden in sudden death in the young.medRxiv [Preprint]. 2023 Mar 29:2023.03.27.23287711. doi: 10.1101/2023.03.27.23287711. medRxiv. 2023. Update in: Genome Med. 2024 Jan 16;16(1):13. doi: 10.1186/s13073-024-01284-w. PMID: 37034657 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
- U01HL131911/HL/NHLBI NIH HHS/United States
- HL128075/HL/NHLBI NIH HHS/United States
- U01 HL131698/HL/NHLBI NIH HHS/United States
- K99 HL168239/HL/NHLBI NIH HHS/United States
- U01 HL131914/HL/NHLBI NIH HHS/United States
- U01 HL131911/HL/NHLBI NIH HHS/United States
- T32 HL144442/HL/NHLBI NIH HHS/United States
- R01 HL128075/HL/NHLBI NIH HHS/United States
- U01HL131914/HL/NHLBI NIH HHS/United States
- 19SFRN34910009/AHA/American Heart Association-American Stroke Association/United States
- U01HL131698/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
